
Manuscript accepted on :23-09-2024
Published online on: 02-10-2024
Plagiarism Check: Yes
Reviewed by: Dr. Manju Jakhar
Second Review by: Dr. Sharad Kamble
Final Approval by: Dr. Anton R Keslav
Muhammad Irfan Bashir 1* , Nur Hidayah Kaz Abdul Aziz 2, Dzul Azri Mohamed Noor 3, Umar Idris Ibrahim1 and Zalina Zahari1
, Nur Hidayah Kaz Abdul Aziz 2, Dzul Azri Mohamed Noor 3, Umar Idris Ibrahim1 and Zalina Zahari1
1Faculty of Pharmacy, Universiti Sultan Zainal Abidin, Besut Campus, Besut, Terengganu, Malaysia
2Discipline of Pharmacology, School of Pharmaceutical Sciences, Universiti Sains Malaysia, Minden, Penang, Malaysia.
3Discipline of Clinical Pharmacy, School of Pharmaceutical Sciences, Universiti Sains Malaysia, Minden, Penang, Malaysia.
Corresponding Author E-mail:irfanbashir@unisza.edu.my
DOI : https://dx.doi.org/10.13005/bpj/2983
Abstract
The aim of the current research was to dock the two abundant bioactive constituents of Polygonum minus leaf extract, in1cluding Quercetin 3-Glucuronide (Miquelianin) and Quercitrin (Quercetin-3-rhamnoside). In-silico Molecular modelling technique was used to predict about a protein (enzyme) interacts with molecules (ligands). Monoamine oxidase-A (MAO-A) is the key enzyme that is involved in the breakdown of neurotransmitters like serotonin and noradrenaline. Drugs that are involved in its inhibition, are considered to be antidepressant agents. This molecular docking study observed the binding energy of selected ligands and their interactions with amino acid residue along with bond types in the MAO-A structure. Molecular docking was done using Molecular Operating Environment (MOE) software, whereas visualization and expression of results were carried out using Discovery Studio (DS) visualizer. Clorgyline was used in this study as a co-crystal ligand, whereas moclobemide was used as a standard MAO-A inhibitor, and Amitriptyline was used as a common antidepressant which also has some MAO-A inhibitory effect. Quercetin 3-glucuronide (Miquelianin) and Quercitrin (Quercetin-3-rhamnoside) have more binding affinities with MAO-A structure as compared to all other drugs. Its interaction pattern was most likely moclobemide and Clorgyline, which are considered standard MAO-A inhibitors in this study. Based on these results, it is concluded that Quercetin 3-Glucuronide (Miquelianin) and Quercitrin (Quercetin-3-rhamnoside) have the potential to become potent MAO-A inhibitors in future.
Keywords
Docking; In-silico; Molecular; MAO-A; Polygonum minus; Quercitrin (Quercetin-3-rhamnoside); Quercetin-3-Glucuronide
Download this article as:| Copy the following to cite this article: Bashir M. I, Aziz N. H. K. A, Noor D. A. M, Ibrahim U. I, Zahari Z. In-silico Molecular Docking of Two Bioactive Constituents (Quercetin 3-Glucuronide and Quercitrin) from Polygonum minus Leaves into Monoamine Oxidase-A Crystal Structure. Biomed Pharmacol J 2024;17(3). |
| Copy the following to cite this URL: Bashir M. I, Aziz N. H. K. A, Noor D. A. M, Ibrahim U. I, Zahari Z. In-silico Molecular Docking of Two Bioactive Constituents (Quercetin 3-Glucuronide and Quercitrin) from Polygonum minus Leaves into Monoamine Oxidase-A Crystal Structure. Biomed Pharmacol J 2024;17(3).Available from: https://bit.ly/3zyulke |
Introduction
Molecular docking is a type of computer modelling that predicts the optimal binding orientation of two molecules one as a ligand and another as a receptor, when they interact to create a complex which is stable 1. The study of bioactive peptides or chemical medicinal compounds that bind to particular receptors is known as in-silico molecular docking. That demonstrates the binding’s shape, pattern and affinity 2. In-silico approaches can locate prospective binding sites and discover and build novel molecules that can bind to a known site. To find novel drugs, blind docking and virtual screening are frequently used 3. Within the molecular docking community, two techniques are particularly prominent. One method employs a matching strategy in which the protein and ligand are described as complementary surfaces 4. The second technique simulates the docking process by computing the pairwise interaction energies between the ligand and the protein. Both systems have several advantages 5. There are some hydrogen bond donors and also the acceptors in the ligand, which are charged, groups. They interact with oppositely charged side chains in the receptor or might be falling into hydrophobic pockets. It can also be checked that hydrophobic groups in the ligand are buried in the receptor’s hydrophobic pockets 6. In docking, root-mean-square deviation, RMSD value is used to compare the docked conformation with the reference confirmation, success is typically regarded if its value is less than 2 Å 7,8 Docking mostly depends on the hydrophobic contacts and hydrogen bonds which could be formed between the proteins and ligand. Its main part is interaction sites, which are located at discrete positions in space suitable for forming the hydrogen bonds or for filling a hydrophobic pocket. Conventional and non-conventional hydrogen bonds, pi-pi bonds and other rotatable bonds are also an important part of the results 9,10. The most likely corresponding intermolecular interactions and binding conformations are identified. The protein backbone is represented as a cartoon. The active site residues and ligand are shown in the stick representation. The water molecule is shown as a white sphere, and hydrogen bonds are shown as dashed lines 11. Another vital part of molecular docking is to determine the amino acid residues, which residues are under the interaction of ligand 12. Monoamine oxidases are important in enzymes in brain that break down the monoamines’ through oxidation by changing their amine group with oxygen. Most cell types in the body have them linked to the outer membrane of mitochondria. Both monoamine oxidase A and B have long been targeted as a vital therapeutic destination for the treatment of the depression and various neurodegenerative diseases. Because of its participation in the modulation of serotonin, MAO-A is found associated with the depression treatment many times 13. These enzymes belong to a flavin-containing amine oxidoreductase protein family14. They play an important role for the inactivation of monoamine neurotransmitters and the breakdown of monoamines present in food. They have been linked with various mental and neurological diseases, which can be treated with the monoamine oxidase inhibitors (MAOIs), which prevent MAOs to perform their functions efficiently 15. MOE software is a molecular modelling program, it is specifically designed to study large biological molecules. This software is also designed to use semi-empirical and ab initio quantum mechanics calculations and different force fields as well 16.
Polygonum minus Huds is a member of the Polygonaceae family and commonly referred to as Kesum or laksa leaf as local name in Malaysia. It is used as a preventive healthcare agent. Most of these herbs are believed to be associated with the anti-oxidant activities and have many beneficial effects 17,18. The leaf part of this herb has been reported to have two major flavonoids like quercetin-3- glucuronide ((Miquelianin) and quercitrin (quercetin-3-rhamnoside) 19.
The current research aimed to dock the two abundant bioactive constituents of Polygonum minus leave extract which are Quercitrin and Quercetin 3-Glucuronide. This molecular docking study was efficiently performed to observe the binding energy of selected ligands and their interactions with amino acid residue along with bond types in Monoamine Oxidase (MAO-A) structure.
Materials and Methods
The docking experiments based on computer aided assistance were carried out by using the crystal structure of MAO-A (PDB ID:2BXR). Docking simulation study of the Bioactive flavonoids of P. minus, Tricyclic antidepressant and MAO-A inhibitors were docked with MAO-A structure. Ligand’s formula structures were derived from the PubChem database. Quercitrin also known as Quercetin-3-rhamnoside (Compound CID 5280459), Quercetin-3-glucuronide (Com-pound CID: 5274585), Clorgyline (Compound CID: 4380) (crystal ligand and MAO-A inhibitor), Moclobemide (Compound CID: 4235) (standard MAO-A inhibitor), Amitriptyline (Compound CID: 2160) (Positive control of current study). Ligands were further prepared and docking simulation was finally done by using MOE dock 2015 software, the following published protocol 20.
Enzyme structures were examined for the missing atoms, bonds and associations.
Then, hydrogen atoms were added to the structure of MAO-A enzyme. Already bound ligands and water molecules were manually deleted.
The structures of ligand molecules were taken from the PubChem database and prepared further for docking through MOE software.
MOE-Alpha Site Finder used to generate the active site.
Dummy atoms were made up from the obtained alpha spheres.
All Ligands were docked within the active site of MAO-A enzyme using the MOE Dock with simulated annealing used as the search protocol.
The least energy conformation was picked and subjected to the minimization of energy.
Finally, the investigation of the 2D and 3D hydrogen-bond linkages was completed using the Biovia Discovery Studio 4.5 program. This program was used to visualise the docking results as an image. This molecular visualization generates graphical images that help in studying the nature of the hydrogen and hydrophobic bonds. This graphical image shows the bond length between interacting atoms of ligands and protein.
Results and Discussion
Molecular docking was performed by using the Molecular Operating Environment (MOE) software. Clorgyline, Moclobemide, Amitriptyline, Quercetin 3-glucuronide (Miquelianin) and Quercitrin (Quercetin-3-rhamnoside) were docked individually into MAO-A prepared structure. Table 1. shows the results of their docking interactions. The lowest binding energy was selected for an individual binding score which determines the high affinity of a ligand with the MAO-A attachment site. RMSD (bond length) was determined by angstrom (Å). All selected values have less than 2 Å RMSD values which are under the range of good docking angle values. This table also explains the residues of amino acid which were interacted with different ligands. Further, each docking interaction has been explained individually.
Table 1: Molecular Docking interactions of Ligands into MAO-A
|
Name of ligand |
Description |
Binding Energy |
Rmsd value |
H-bonding |
Hydrophobic Interactions |
||
|
Conventional |
Carbon-hydrogen |
Alkyl and Pi-alkyl |
Others |
||||
|
Amitriptyline |
Positive control Antidepressant |
-7.8829 |
1.52 |
– |
GLY A:443 TYR A:407 |
LYS A:305, MET A:445, VAL A:303 |
CYS A:406 (π-sulfur), GLY A:67 (Vander walls), GLY A:66 (π- π stacked), TRP A:397 (amide- π stacked) |
|
Clorgyline |
Co crystal ligand -re-dock MAO-A inhibitor |
-7.2970 |
1.47 |
– |
GLN A:215, GLY A:66 |
CYS A:323, ILE A:180, ILE A:335, LEU A:337, LYS A:305, PHE A:352, TYR A:407, TYR A:69 |
– |
|
Moclobemide |
Standard MAO-A inhibitor |
-7.0720 |
1.31 |
– |
GLY A:443, TYR A:69 |
ILE A:180, ILE A:335, MET A:445, TYR A:444 |
– |
|
Quercitrin (Quercetin-3-rhamnoside) |
Bioactive constituent of P. minus |
-8.5182 |
1.15 |
GLN A:215, CYS A:406, GLY A:67, ARG A:51 MET A:445 |
PHE A:352, TYR A:444 GLY A:443 |
TYR A:69 |
TRP A:397 (π-π T-shape) |
|
Quercetin-3-glucuronide |
Bioactive constituent of P. minus |
-8.3633 |
1.29 |
GLY A:443, TYR A:197, MET A:445, ALA A:68, TYR A: 69 |
GLY A: 67, TYR A:444 |
ILE A:335, ILE A:480, |
TYR A:407 (π- π stacked) |
Docking of Amitriptyline into MAO-A
Amitriptyline showed the binding energy of -7.8828 kcal/mol which exhibits the good inhibitory potential of a compound. Detailed molecular interaction pattern of amitriptyline docked pose demonstrated that one of the phenyl ring A and aliphatic nitrogen is involved in the formation of carbon-hydrogen bonding with TYR A: 407 and GLY A:443 amino acid residues. Substituted methyl group and phenyl ring B establish alkyl and π-alkyl interaction with LYS A:305, MET A:445, VAL A:303 and respectively. The hydrophobic cleft formed by GLY A:67 (Vander walls), GLY A:66 (π- π stacked), and TRP A:397 (amide- π stacked) provides additional stability to a ligand. Apart from this CYS A:406 (π-sulphur linkages) contributes to the high binding affinity of a ligand with MAO-A (Figure 1).
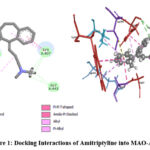 |
Figure 1: Docking Interactions of Amitriptyline into MAO-A |
Docking of Clorgyline into MAO-A
Clorgyline having the binding energy -7.2970 kcal/mol was found to be positioned into the binding pocket assembled by CYS A:323, ILE A:180, ILE A:335, LEU A:337, LYS A:305, PHE A:352, TYR A:407, TYR A:69 amino acid residues. Moreover, the docked complex between Clorgyline and MAO-A was stabilized by hydrogen bond interactions between the aliphatic hydrogen of the ligand and the chains of GLN A:215 and GLY A:66 amino acids (Figure 2).
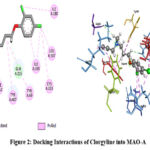 |
Figure 2: Docking Interactions of Clorgyline into MAO-A |
Docking of Moclobemide into MAO-A
Moclobemide showed binding energy of -7.0719 kcal/mol and exhibits the good inhibitory potential of the compounds. Detailed molecular interaction pattern of Moclobemide docked pose demonstrated that aliphatic hydrogen was involved in the formation of hydrogen bonding with GLY A:443 and TYR A:69 amino acid residues. Substituted chloride group and morpholine ring established an alkyl and π-alkyl interaction with MET A:445, TYR A:444, and ILE A:180, ILE A:335 residues respectively (Figure 3).
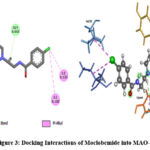 |
Figure 3: Docking Interactions of Moclobemide into MAO-A |
Docking of Quercitrin (Quercetin-3-rhamnoside) into MAO-A
Quercitrin (Quercetin-3-rhamnoside) showed binding energy of -8.5182 kcal/mol that exhibits the good inhibitory potential of the compounds. Detailed molecular interaction pattern of LIGAND docked pose demonstrated that GLN A:215, CYS A:406, GLY A:67, ARG A:51 and MET A:445 amino acid residues are involved in the formation of Conventional hydrogen bonds. Whereas, PHE A:352, TYR A:444 and GLY A:443 are forming a Carbon hydrogen bond. Substituted phenyl ring forms π-π T-shape interaction with TRP A:397 and aliphatic methyl forms π-alkyl interaction with TYR A:69 amino acid residues respectively (Figure 4).
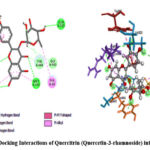 |
Figure 4: Docking Interactions of Quercitrin (Quercetin-3-rhamnoside) into MAO-A |
Docking of Quercetin 3-glucuronide into MAO-A
Quercetin-3-glucuronide showed -8.3633 kcal/mol binding energy which indicates a higher inhibitory effect as compared to all other docked ligands in this study. Querce-tin-3-glucuronide formed 7 hydrogen bonds with GLY A:443, TYR A:197, MET A:445, ALA A:68, TYR A: 69 (conventional), and GLY A: 67, TYR A:444 (Carbon-hydrogen) respectively. It also forms hydrophobic alkyl and π-alkyl interactions with ILE A:335, and ILE A:480 respectively. Moreover, docked complex between Quercetin-3-glucuronide and protein was stabilized by π-π stacked interactions between the phenyl ring of the ligand and the side chain of TYR A:407 amino acid (Figure 5).
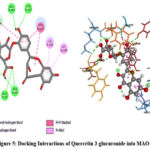 |
Figure 5: Docking Interactions of Quercetin 3-glucuronide into MAO-A |
Amongst all docked ligands, Quercitrin (Quercetin-3-rhamnoside) and Quercetin-3-glucuronide showed the higher binding energy-8.5182 kcal/mol and -8.3633 kcal/mol respectively as compared to other MAO-A inhibitors (standard drugs). and both were well oriented into the binding pocket of MAO-A as well. As shown in Table 1 Q3G and quercitrin had some common amino acid residue interactions with moclobemide and clorgyline. They possessed some similarities to standard MAOIs but with higher binding affinity. Table 2 expresses the common amino acid residue interactions between different ligands.
Table 2: Common amino acid residue interactions between different pairs of ligands.
|
Ligand Pairs |
Common Interactions |
|
Moclobemide and Clorgyline |
TYR A:69, ILE A:335, ILE A:180, |
|
Moclobemide and Quercetin-3-glucuronide |
TYR A:69, GLY A:443, ILE A:335, Tyr A:444, ILE A:335 MET A:445 |
|
Moclobemide and Quercitrin (Quercetin-3-rhamnoside)
|
TYR A:69, GLY A:443, MET A:445, TYR A:444 |
|
Clorgyline and Quercetin-3-glucuronide |
TYR A:69, ILE A:335, TYR A:407 |
|
Clorgyline and Quercitrin (Quercetin-3-rhamnoside) |
TYR A:69, GLN A:215, PHE A:352 |
|
Moclobemide, Clorgyline Quercetin-3-glucuronide and Quercitrin, |
TYR A:69 |
|
Moclobemide, Clorgyline Quercetin-3-glucuronide |
TYR A:69, GLY A:443 |
|
Moclobemide, Clorgyline and Quercitrin (Quercetin-3-rhamnoside) |
TYR A:69, ILE A:335 |
|
Quercetin-3-glucuronide and Quercitrin (Quercetin-3-rhamnoside) |
TYR A: 69, GLY A:443, MET A:445, GLY A: 67, TYR A:444 |
|
Moclobemide and Amitriptyline |
GLY A:443, MET A:445 |
|
Clorgyline and Amitriptyline |
GLY A:66, TYR A:407 |
|
Moclobemide, Clorgyline and Amitriptyline |
———– |
|
Amitriptyline and Quercetin-3-glucuronide |
GLY A:443, TYR A:407, MET A:445, GLY A:67 |
|
Amitriptyline and Quercitrin (Quercetin-3-rhamnoside) |
GLY A:443, MET A:445 GLY A:67, TRP A:397 |
The current study is providing the results regarding molecular docking of Quercitrin (Quercetin-3-rhamnoside) and Quercetin-3-glucuronide with MAO-A structure first time in literature, it showed that these constituents have many similar amino acid residues with standard MAO-A inhibitors but there are no any previous studies on docking of these constituents for their comparison.
Moclobemide is selective reversible inhibitor of MAO-A, it increases the monoamines level inside the brain. Moclobemide was used as a standard MAO-A inhibitor in this docking study. It showed similar interactions by using MOE software in the present study in comparison to the latest performed research on Moclobemide and MAO-A docking examination carried out by using Auto Dock 4 software. In which amino acid residues like GLY A:443, TYR A:69, ILE A:180, ILE A:335, MET A:445 and TYR A:444 were similar 21.
Clorgyline was used as a co-crystal ligand in this study. The mechanism of interaction of reversible monoamine oxidase (MAO)‐A inhibitor with monoamine oxidase Its interactions with CYS A:323, ILE A:180, ILE A:335, LEU A:337, LYS A:305, PHE A:352, TYR A:407 and TYR A:69 were quite similar to previously done research work regarding MAO-A inhibition through molecular docking, but in that study, researchers used Auto dock 3.0 software instead of MOE software 22.
Amitriptyline also showed some interactions with TYR A:407, GLY A:443, LYS A:305, MET A:445, VAL A:303, CYS A:406 (π-sulphur), GLY A:67 (Vander walls), GLY A:66 (π- π stacked), TRP A:397 (amide- π stacked) like other MAO-A inhibitors, which confirms that it also has some MAO-A inhibition activity, before this previous research has al-ready demonstrate that although it is a tricyclic antidepressant it has also inhibitory effect on MAO-A 23.
The current molecular study suggested that Quercitrin (Quercetin-3-rhamnoside) and Quercetin-3-glucuronide, both ligands may have the capability to act like moclobemide and clorgyline with more binding affinity with MAO-A structure as compare to these standard ligands. This work also recommended that P. minus aqueous leaf extract may have an MAO-A inhibitory effect due to the presence of these two bioactive constituents.
Conclusion
Quercetin 3-glucuronide (Miquelianin) and Quercitrin (Quercetin-3-rhamnoside) have more binding affinity with MAO-A structure as compared to standard MAO inhibitors like Clorgyline, Moclobemide and Amitriptyline (Tricyclic antidepressants). Based on a Molecular docking study, Quercetin 3-glucuronide (Miquelianin) and Quercitrin (Quercetin-3-rhamnoside) may responsible for MAO-A inhibition activity of P. minus leaf because they have several same bonding interactions with amino acid residues of MAO-A enzyme, like other standard MAO-A inhibitors. Quercetin 3-glucuronide (Miquelianin) and Quercitrin (Quercetin-3-rhamnoside) can be proved to be a potent MAO-A inhibitor substance in future.
Acknowledgement
The author acknowledge the Biovia software company to provide the free software.
Conflict of Interest
The author(s) do not have any conflict of interest
Funding Source
The author(s) received no financial support for the research.
Data Availability Statement
The manuscript incorporates all datasets produced or examined throughout this research study and can be accessed easily on request
Ethic Statement
This research did not involve human participants, animal subjects, or any material that requires ethical approval
Informed Consent Statement
This study did not involve human participants, and therefore, informed consent was not required.
Authors’ Contributions
Muhammad Irfan Bashir: Performed the molecular docking and conceptualized as well as designed the experiment.
Nur Hidayah Kaz Abdul Aziz and Dzul Azri Mohamed Noor: conceptualized and design the experiments.
Muhammad Irfan Bashir and Nur Hidayah Kaz Abdul Aziz: wrote the article.
Umar Idris Ibrahim: visualization
Zalina Zahri: review and editing.
References
- Agarwal S, Mehrotra R. An overview of Molecular Docking. JSM Chem. 2016;4(2):1024-1028.
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Reviews Drug Discovery. 2004;3(11):935-949.
CrossRef - Rao V, Srinivas K. Modern drug discovery process: An in silico approach. Journal of Bioinformatics and Sequence Analysis. 2011;2(5):89-94.
- Zhong S, Zhang Y, Xiu Z. Rescoring ligand docking poses. Current Opinion in Drug Discovery & Development. 2010;13(3):326-334.
- Islam M. Molecular Docking an Important Tool for Drug Designing. 2018;1(4):1-9. doi:10.31031/MADD.2018.01.000518
CrossRef - Morris GM, Lim-Wilby M, Kukol A. Molecular Docking. Humana Press.:365-382. doi:10.1007/978-1-59745-177-2_19
CrossRef - Gohlke H, Hendlich M, Klebe G. Predicting binding modes, binding affinities and hot spots’ for protein-ligand complexes using a knowledge-based scoring function. Perspectives in Drug Discovery and Design. 2000;20(1):115-144. doi:10.1023/A:1008781006867
CrossRef - Yusuf D, Davis AM, Kleywegt GJ, Schmitt S. An alternative method for the evaluation of docking performance: RSR vs RMSD. Journal of Chemical Information and Modeling. 48(7):1411-1422. doi:10.1021/CI800084X
CrossRef - Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Current computer-aided drug design. 2011;7(2):146-157.
CrossRef - Bitencourt-Ferreira G, Veit-Acosta M, de Azevedo WF. Hydrogen Bonds in Protein-Ligand Complexes. Methods in Molecular Biology (Clifton, NJ). 2019;2053:93-107
CrossRef - Ferreira L, dos Santos R, Oliva G, Andricopulo A. Molecular Docking and Structure-Based Drug Design Strategies. Molecules. 2015;20(7):13384-13421.
CrossRef - Jing Z, Feng H. Studies on the Molecular Docking and Amino Acid Residues Involving in Recognition of Substrate in Proline Iminopeptidase by Site-Directed Mutagenesis. The Protein Journal. 2015;34(3):173-180.
CrossRef - Mathew B, E. Mathew G, Suresh J, Ucar G, Sasidharan R, Anbazhagan S, Vilapurathu J, Jayaparkash V. Monoamine Oxidase Inhibitors: Perspective Design for the Treatment of Depression and Neurological Disorders. Current Enzyme Inhibition. 2016;12(2):115-122.
CrossRef - Harismah K, Mirzaei M. In silico interactions of steviol with monoamine oxidase enzymes. Lab-in-Silico. 2020;1(1): 3-6.
- Yeung AWK, Georgieva MG, Atanasov AG, Tzvetkov NT. Monoamine Oxidases (MAOs) as Privileged Molecular Targets in Neuroscience: Research Literature Analysis. Frontiers in Molecular Neuroscience. 2019;12(1):1-35.
CrossRef - Chan PF, Srikannathasan V, Huang J, Cui H, Fosberry AP, Gu M, Hann MM, Hibbs M, Homes P, Ingraham K, Pizzollo J, Shen C, Shillings AJ, Spitzgaden CE, Tanner R, Theobald AJ, Stavenger RA, Bax BD, Gwyn MN. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nature Communications. 2015;6(1):1-13.
CrossRef - Christapher P, Parasuraman S, Christina JA, Vikneswaran M, Asmawi MohdZ. Review on Polygonum minus. Huds, a commonly used food additive in Southeast Asia. Pharmacognosy Research. 2015;7(1):1-6.
CrossRef - Nordin NA, Ibrahim AR, Ngaini Z. Biological Studies of Novel Aspirin-Chalcone Derivatives bearing Variable Substituents. Journal Of Agrobiotechnology. 2020;11(1):20-31.
CrossRef - Lau H, Shahar S, Mohamad M, Rajab N, Yahya H, Che N, Abdulhamid H. The effects of six months Persicaria minor extract supplement among older adults with mild cognitive impairment: a double-blinded, randomized, and placebo-controlled trial. BMC Complementary Medicine and Therapies. 2020;20(1).
CrossRef - Khattab SN, Hassan SY, Bekhit AA, Abdel, Langer V, Amer A. Synthesis of new series of quinoxaline-based MAO-inhibitors and docking studies. European journal of medicinal chemistry. 2010;45(10):4479-4489.
CrossRef - Alidoosti ZS, Mirzaei M. Comparative Examination of Moclobemide, Tranylcypromine, Phenelzine and Isocarboxazid for Monoamine Oxidase–A.
- Tao G, Irie Y, Li DJ, Keung WM. Eugenol and its structural analogs inhibit monoamine oxidase A and exhibit antidepressant-like activity. Bioorganic & Medicinal Chemistry. 2005;13(15):4777-4788.
CrossRef - Fisar Z, Hroudova J, Raboch J. Inhibition of monoamine oxidase activity by antidepressants and mood stabilizers. Neuro endocrinology letters. 2023;31(5):645-656.

