
Manuscript accepted on :26-08-2024
Published online on: 24-09-2024
Plagiarism Check: Yes
Reviewed by: Dr. Ahmed Falyyih
Second Review by: Dr. Anjaneyulu Vinukonda
Final Approval by: Dr. Prabhishek Singh
Nasir Abdelrafie Hamad1* , Lienda Bashier Eltayeb2 and Habab Merghani Yassin3
, Lienda Bashier Eltayeb2 and Habab Merghani Yassin3
1Department of Biochemistry, College of Medicine and Health Sciences, National University of Sciences and Technology, Sohar Oman. College of Medicine, El-Neelain University, Sudan.
2Department of Medical Laboratory Sciences, College of Applied Medical Sciences, Prince Sattam bin Abdulaziz University, Al-Kharj, Riyadh, Saudi Arabia.
3Department of Biology, Faculty of Science, King Khalid University, Abha, Saudi Arabia.
Corresponding Author E-mail:nasircosha@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/2996
Abstract
Background: Early-onset Alzheimer's disease (EOAD) constitutes 1-2% of all Alzheimer's cases, presenting with poorer prognosis, progressive symptoms, and reduced life expectancy compared to late-onset Alzheimer’s, thereby increasing socioeconomic burden. Elevated plasma homocysteine levels due to MTHFR gene polymorphisms are implicated in Alzheimer's etiology. The present study aims to explore the association between MTHFR gene polymorphisms in Sudanese population. Methods: Seventy-three EOAD patients were assessed for MTHFR C677T and A1298C polymorphisms, alongside plasma homocysteine levels. Results: Significant associations were observed between CT and TT alleles, elevated plasma homocysteine levels, and EOAD. Conclusion: MTHFR C677T polymorphism was associated in EOAD in Sudanese population. Elevated plasma homocysteine levels might frame this association and potentially contribute to the disease onset before the age of 65.
Keywords
A1298C, C677T; Allele; Alzheimer's disease, Early-onset; Heterozygous; Homozygous; MTHFR, Polymorphism
Download this article as:| Copy the following to cite this article: Hamad N. A, Eltayeb L. B, Yassin H. M, MTHFR Polymorphisms and Plasma Homocysteine in Early-Onset Alzheimer's Disease: A Case-Control Study. Biomed Pharmacol J 2024;17(3). |
| Copy the following to cite this URL: Hamad N. A, Eltayeb L. B, Yassin H. M, MTHFR Polymorphisms and Plasma Homocysteine in Early-Onset Alzheimer's Disease: A Case-Control Study. Biomed Pharmacol J 2024;17(3). Available from: https://bit.ly/3ZAuo9t |
Introduction
Incidence
Alzheimer’s disease (AD) is an age-accelerated progressive neurodegenerative disorder characterized by a decline in both behavioural and cognitive functions1,2. The disease is the most common type of dementia in individuals aged 65 years or above2,3, accounting for 50-70% of all cases4. Globally, dementia accounted for 3.9 % in people aged 65 years or above5. Regionally, the lowest prevalence of dementia was reported in Africa (1.6%), followed by China and Western Pacific regions (4.0%). In Latin America, it accounted for 4.6% of cases, The highest prevalence was reported in North America (6-4%) and Western Europe (5.4%)5.
Alzheimer’s disease is classified as early-onset Alzheimer’s disease (EOAD) and late-onset Alzheimer’s disease (LOAD); based to the onset of the disease under or over 65 years old6, respectively. About 10-30% of individuals aged above 65 years develop late-onset Alzheimer’s disease7. Early-onset Alzheimer’s disease, on the other side, represented 5-10% of all cases of AD8. In other studies, however, the prevalence was as low as 3%9. The mean age for AD patients varied among different populations. In one study, the mean age was 72 ± 5 years for LOAD and 60 ± 4 years for EOAD10. In another study, the mean age for LOAD and EOAD was 74 ± 6 years and 56 ± 5 years, respectively11.
Literature has already established the correlation between gender and AD, with highest rates detected in women than men12. This gender-related variation was attributed to hormonal and psychological factors12. In an attempt to find an explanation, some studies have suggested a protective role of the female sex hormone oestrogen against neuronal degeneration, and hence postmenopausal women have a greater risk of AD13.
Clinically, Alzheimer’s disease is characterized by a progressive decline in episodic memory and cognitive function parameters such as language, attention, reasoning, memory, comprehension, judgement, execution, and visuospatial abilities14. The disease has three stages which take place in a chronological manner. The first stage is called the preclinical or asymptomatic stage, owing to the asymptomatic nature of the stage. The second stage is the cognitive impairment stage, which is followed by the third stage of dementia2. It is worth mentioning that the clinical presentation of early-onset and late-onset disease is almost the same14. A notable difference is that patients with the early-onset disease tend to preserve the episodic memory function rather than the cognitive function14. Additionally, patients of the early-onset disease tended to show a more aggressive course with more frequent pathological patterns and a relatively shorter life span14.
Generally, AD is diagnosed on clinical bases as the disease pathology can be seen in otherwise asymptomatic individuals. Moreover, disease biomarkers like plasma and cerebrospinal fluid markers2 were unable to detect all abnormalities in amyloid peptide and tau protein15. The clinical criteria for the diagnosis of dementia and Alzheimer’s disease were both implemented in the diagnosis of AD16.
The aetiology of Alzheimer’s disease is not fully understood16 and is thought to have a multifactorial nature17. This is applied, in particular; to cases of LOAD18. The main pathological abnormalities in AD included the extracellular accumulation of amyloid-beta (Aβ) plaques and the intraneuronal deposition of tau proteins (p-tau)19. Other abnormalities included the synaptic and neuronal loss20, the vascular dysfunction9, and the brain atrophy21. Furthermore, infection22, genetic susceptibility23, and mutations23 were all incriminated in the disease pathology. Other factors included abnormal acetylcholine in the brain, insulin resistance, vascular dysfunction, oxidative stress, mitochondrial dysfunction and inflammation9. In addition, several risk factors for the disease were identified, including advanced age18, ethnicity24, diabetes9, Down syndrome9, cardiovascular and cerebral diseases9, environmental factors9, smoking25 and alcohol consumption25.
Genetically, the ε4 allele of the APOE gene is the most common identified mutation associated with the late onset disease8. For the early-onset disease, almost all cases have familial nature9, with 30-60% of cases have at least one affected first-degree relative26, and 10% have autosomal dominant pattern of inheritance8. The familial pattern of the disease was attributed to mutations affecting Aβ precursor protein (APP) gene, Presenilin 1 (PSEN1) gene, and Presenilin 2 (PSEN2) gene9. Those genes are affected by over 400 known mutations8.
Pathologically, the mutations are thought to increase not only the production of β-amyloid proteins23 but also its deposition8. Other suggested mutations include those affecting APOE, BIN1, SORL1, CLU, TREM29, and ABCA727 genes. The methylene tetrahydrofolate reductase (MTHFR) gene has three polymorphisms associated with AD: C677T, A1298C, and A1793G28. MTHFR C677T was reported as the most significant of these mutations28. Regarding the other two polymorphisms, it was hypothesized that they protect against the development of AD28. Furthermore, MTHFR C677T was proposed as a risk factor for AD in a study on Asians29, and in a meta-analysis study30.
Both the heterozygous (CT) and homozygous alleles (TT) of MTHFR C677T polymorphism were associated with an increased risk for AD31. Interestingly, a study on Chinese population concluded that both MTHFR C677T and A1298C polymorphisms were associated with the late onset disease28. This conclusion was supported by a case study which detected both C677T and A1298C polymorphisms in a patient diagnosed with early-onset Alzheimer’s disease32. Moreover, another study found that A1298C polymorphism, but not MTHFR C677T; was associated with the disease33. Among the theories that explained the role of MTHFR C677T polymorphism in the development of AD were the white matter signal abnormalities caused by hyperhomocysteinemia34, the impaired function and deposition of the neuronal amyloid-β protein precursor (AβPP) caused by the polymorphism-induced phosphorylation of amyloid- β protein precursor35, and the impaired cognitive function caused by the low folate levels secondary to MTHFR C677T polymorphism36.
Methylene tetrahydrofolate reductase (MTHFR) is an enzyme that catalyses the biochemical conversion of 5,10-methylenetetrahydrofolate to 5-methyltrahydrofolate37. The 5-methyltetrahydrofolate is a coenzyme in the formation of methionine from homocysteine37. The gene encoding the enzyme is located on the short arm of chromosome 1 (1p36.3)37. More than 40 single nucleotide polymorphisms in MTHFR gene were described38, the most common being C677T and A1298C39. MTHFR C677T polymorphism results in the formation of a thermolabile enzyme40. Accordingly, enzyme activity will be diminished, resulting in elevated levels of homocysteine, a condition known as hyperhomocysteinemia41. MTHFR C677T polymorphism and the resulting hyperhomocysteinemia were significantly associated with the development of several diseases and syndromes including subacute combined degeneration of the spinal cord42, cardiac syndrome X43, hypertrophic cardiomyopathy44, cleft lip/palate45, β-thalassemia46, preeclampsia47, diabetes mellitus48, rectal cancer49, stroke50, neural tube defects51, psoriasis52, late-onset Alzheimer’s53, and bipolar disorders54. Literature has reported a global frequency of MTHFR 677CT of about 25%. Regionally, the highest frequency was 47%, reported among Hispanics. The frequency dropped to 36% in Europeans, 30% in East Asians, 12% in South Asians, and finally 9% among Africans55. Additionally, the frequency of the homozygous and heterozygous alleles in the general population was 8.5% and 2.5%, respectively56.
Currently, there is no treatment available for AD19. The life expectancy of the disease depends on age, gender, and genetics19 and is relatively shorter in EOAD than LOAD14. Furthermore, the early-onset disease has a poorer prognosis than the late-onset disease as its course is more progressive14. Accordingly, EOAD has a greater socioeconomic burden14 given that the associated responsibilities like raising children and working53. Moreover, EOAD is associated with a considerable delay in diagnosis54, reflected as higher rates of premature mortality6.
Materials and Methods
Study Type and Population
This case-control study was conducted among Alzheimer’s patients in Khartoum, Sudan, during the period from May 2021-May 2022. Participants were recruited from different neuropsychiatric and healthcare facilities in Khartoum. The sample size was calculated based on a 95% confidence interval, a 5% margin of error, an estimated 5% population proportion, and a population size of 6000 citizens, assuming a normal sample distribution57. The population proportion was taken as 5% based on previous well-designed review study8. The following formula was used to calculate the sample size58:
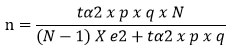
whereas:
n = sample size
N = population size
p = expected percentage of the variable
q = 1-p
e = accepted margin of error
tα = 1.96 for 95% confidence interval.
Accordingly, a total of 73 patients; clinically diagnosed with EOAD, were enrolled using simple random sampling. All patients diagnosed by expert neurologists and met the clinical criteria for diagnosis of dementia15, and AD clinical stages15 during the study period were included in the study. The control group included 73 healthy subjects recruited from individuals attending the same facilities for other reasons.
Data Collection
Sociodemographic data and medical data related to Alzheimer’s disease were collected from the hospital’s medical records after obtaining an ethical approval.
DNA Extraction and PCR
Whole blood was collected, and DNA was extracted using Qiagen kits (FlexiGene DNA Kit)59. DNA was amplified by polymerase chain reaction using GeneAmp PCR kit (PerkinElmer Cetus)60. The 198 bp amplified fragments were digested with HinfI endonuclease which identifies C-T substitution. Accordingly, fragments were cut into 175 and 23 bp fragments. Fragments were then electrophoresed in polyacrylamide gel and stained with ethidium bromide43. The wildtype (CC) allele appeared as a 198 bp band whereas the homozygous (TT) allele appeared as a 175 bp band. The heterozygous (CT) allele appeared as two bands (198 and 175 bp)43. For A1298C polymorphism, the amplified 163 bp was digested with MboII. The AA genotype produced 5 fragments of 56, 31, 30, 28 and 18 bp. The homozygous (CC) allele gives 4 fragments of 84, 31, 30 and 18 bp whereas the heterozygous (AC) allele produced 6 fragments of 84, 56, 31, 30, 28 and 18 bp43.
Plasma Homocysteine
Plasma homocysteine level was measured using Human Homocysteine (HCY) ELISA Kit61 . EDTA tube was used to collect blood. sample was immediately Centrifuged for 15 minutes at 1000 x g, 2-8°C. Plasma homocysteine levels over 15 micromole/l were considered as hyperhomocysteinemia62.
Data Presentation and Analysis
Descriptive data were presented as means and standard deviations for quantitative variables and frequencies for qualitative variables. Results were statistically analysed using the SPSS (18th version). Chi-Square test was used to compare gender and the distribution of MTHFR C677T genotypes between EOAD patients and controls. The two-tailed t-test was used to compare means of plasma homocysteine between AD cases and control subjects. Analysis of variance (ANOVA) test was used to correlate plasma homocysteine with the distribution of MTHFR C677T genotypes. The p-value was considered significant when ≤ 0.05.
Ethical approval
The study was ethically approved by the local ethical committee at Faculty of Medicine, El-Neelain University, and the Ministry of Health, Sudan.
Results
Demographic Data
The current study recruited 73 Sudanese patients with early-onset Alzheimer’s and other 73 control subjects. The mean age of participants was 57.9 years (table 1). With regards to gender, males represented 27.3% and 53 females represented 72.4% (table 2). The difference between cases and control subjects was significant (p=0.000).
Table 1: Mean age of EAOD patients
|
Count, n |
73 |
|
Sum, Σx |
4221 |
|
Mean, x̄ |
57.821917808219 |
|
Variance, s2 |
9.9539573820396 |
|
Standard deviation |
3.1549892839817 |
|
Standard error of mean (SEM) |
0.36926356518872 |
|
Confidence of interval |
95%, 1.960sx̄ |
|
Margin of error (confidence interval 95%) |
57.8219 ±0.724 (±1.25%) |
Table 2: Sex distribution among EOAD cases and controls
|
Cases |
Observed |
20 (27.4%) |
53 (72.6%) |
73 |
df = 1 |
|
|
Expected |
33 |
40 |
|
p-value = 0.000 |
|
|
Chi Square contribution |
5.1212 |
4.225 |
|
Chi Square = 18.6924 |
|
Controls |
Observed |
46 () |
27 |
73 |
|
|
|
Expected |
33 |
40 |
|
|
|
|
Chi Square contribution |
5.1212 |
4.225 |
|
|
|
Column totals |
|
66 |
80 |
146 |
|
Plasma Homocysteine
Table 3 shows that the mean plasma homocysteine level was 15.5 micromole/l among cases and 13.01 micromole/l among control subjects. The difference in the mean plasma homocysteine level between cases and control subjects was significant (p=0.0001).
Table 3: Mean plasma homocysteine levels among EOAD and controls
|
AD |
Count, N: |
73 |
df = 144 Standard error of difference = 0.172 t = 14.221F two-tailed p value = 0.0001 95% CI = 2.1112572999529675-2.792852 2890890335. |
|
|
Sum, Σx: |
1129 |
|
|
|
Mean, x̄: |
15.465753424658 |
|
|
|
Variance, s2: |
1.8356164383562 |
|
|
|
Standard deviation |
1.3548492308579 |
|
|
|
Standard error of mean (SEM) |
0.1585731082574 |
|
|
|
Confidence of interval |
95%, 1.960sx̄ |
|
|
|
Margin of error |
15.4658 ±0.311 (±2.01%) |
|
|
CONTROL |
Count, N: |
73 |
|
|
|
Sum, Σx: |
950 |
|
|
|
Mean, x̄: |
13.013698630137 |
|
|
|
Variance, s2: |
0.33453196347032 |
|
|
|
Standard deviation |
0.57838738183878 |
|
|
|
Standard error of mean (SEM) |
0.067695122694177 |
|
|
|
Confidence of interval |
95%, 1.960sx̄ |
|
|
|
Margin of error |
13.0137 ±0.133 (±1.02%) |
MTHFR C677T Genotypes
Regarding MTHFR C677T genotype distribution among cases of EOAD, we found that CC, CT, and TT frequencies were 24.7%, 27.4% and 47.9% respectively. Genotype frequencies among control subjects were 78%, 12.3%, and 9.6%, respectively (table 4). The difference in genotype frequencies between cases and control subjects was statistically significant (p=0.0001). Table 5 shows the mean age among different MTHFR C677T genotypes of EOAD patients. Statistical analysis was significant (p= 0.000). The gender distribution of MTHFR C677T genotypes among cases of EOAD was shown in table 6. The difference among MTHFR C677T genotypes was significant (p= 0.000). Moreover, we compared the plasma homocysteine levels among MTHFR C677T genotypes (table 7). Statistical analysis found the difference between genotypes to be significant (p= 0.000).
Table 4: MTHFR C677T genotype distribution among EOAD cases and controls
|
|
|
CT |
TT |
CC |
Raw Totals |
|
|
Cases |
Observed |
20 (27.4%) |
35 (47.9%) |
18 (24.7%) |
73 |
df = 2 |
|
|
Expected |
14.5 |
21 |
37.5 |
|
p-value = 0.0001 |
|
|
Chi Square contribution |
2.0862 |
9.3333 |
10.14 |
|
Chi Square = 43.1191 |
|
Controls |
Observed |
9 (12.3%) |
7 (9.6%) |
57 (78%) |
73 |
|
|
|
Expected |
14.5 |
21 |
37.5 |
|
|
|
|
Chi Square contribution |
2.0862 |
9.3333 |
10.14 |
|
|
|
Column Totals |
|
29 |
42 |
75 |
146 |
|
Table 5: Mean age among MTHFR C677T genotypes of cases
|
Group |
n |
Mean age |
SD |
|
|
CT |
20 |
57.95 |
0.6863 |
|
|
TT |
35 |
57.5143 |
0.7425 |
|
|
CC |
18 |
59.0556 |
0.8726 |
|
|
|
df |
SS |
F-statistic |
p-value |
|
Variation among samples |
2 |
28 |
24.4160 |
0.000 |
|
Variation within samples |
70 |
42 |
||
|
Total |
72 |
69 |
Table 6: Gender distribution of MTHFR C677T genotypes among cases
|
Males |
Observed |
2 (10%) |
5 (25%) |
13 (65%) |
20 |
df = 2 |
|
|
Expected |
5.4795 |
9.5890 |
4.9315 |
|
p-value = 0.000 |
|
|
Chi Square contribution |
2.2095 |
2.1962 |
13.2010 |
|
Chi Square = 25.2506 |
|
females |
Observed |
18 (33.9%) |
13 (24.5%) |
5 (9.4%) |
53 |
|
|
|
Expected |
14.5205 |
25.4110 |
13.0685 |
|
|
|
|
Chi Square contribution |
0.8338 |
0.8287 |
4.9815 |
|
|
|
|
Column Totals |
20 |
35 |
18 |
73 |
|
Table 7: Mean plasma homocysteine levels among MTHFR C677T genotypes
|
CT |
20 |
15.1 |
(14.8565, 15.3435) |
|
|
TT |
35 |
15.5714 |
(15.3874, 15.7555) |
|
|
CC |
18 |
13.8333 |
(13.5767, 14.09) |
|
|
|
df |
SS |
F-statistic |
p-value |
|
Variation among samples |
2 |
36 |
60.5622 |
0.000 |
|
Variation within samples |
70 |
21 |
||
|
Total |
72 |
57 |
MTHFR A1298C Genotypes
Regarding MTHFR 1298C, we reported frequencies of 38.4%, 27.4%, and 34.2% for AA, AC, and CC genotypes, respectively. Genotype frequencies among control subjects were 35.6%, 32.9%, and 31.5%, respectively (table 8). There was no statistical difference between cases and control subjects (p= 0.6228).
Table 8: MTHFR A1298C genotype distribution among EOAD cases and controls
|
Cases |
Observed |
20 (27.4%) |
25 (34.2%) |
28 (38.4%) |
73 |
df = 3 |
|
|
Expected |
22 |
24 |
27 |
|
p-value = 0.6228 |
|
|
Chi Square contribution |
0.1818 |
0.0417 |
0.037 |
|
Chi Square = 1.764 |
|
Controls |
Observed |
24 (32.9%) |
23 (31.5%) |
26 (35.6%) |
73 |
|
|
|
Expected |
22 |
24 |
27 |
|
|
|
|
Chi Square contribution |
0.1818 |
0.0417 |
0.037 |
|
|
|
|
Column totals |
44 |
48 |
54 |
146 |
|
Discussion
In the current study, we explored the relationship between MTHFR C677T and A1298C polymorphisms and early (young)-onset Alzheimer’s disease among Sudanese population. We recruited seventy-three subjects clinically diagnosed with EOAD who were attending or admitted to different neuropsychiatric healthcare facilities in Khartoum, Sudan. EOAD is defined as dementia that occurs before the age 65 years6. This entity of AD is of particular clinical and socioeconomic importance owing to its poor prognosis, early presentation, and tremendous consequences on life quality14. Clinically, EOAD differs from LOAD in some respects. Firstly, EOAD tends to preserve the episodic memory function but not the cognitive impairment14. Secondly, EOAD tends to have more aggressive course and relatively shorter life span than LOAD14. Thirdly, the disease is often underestimated and misdiagnosed54, which was reflected as relatively higher mortality rates6. The global frequency of EOAD was 24.2 in 100000 (0.2%)6. Despite the low frequency, EOAD is considered was considered as the most common dementia before the age 65 years6. Because the study belonged to the case-control entity, we were not able to compute the prevalence of EOAD in Sudanese population.
The mean age of EOAD patients in our study was 57.9 ±3 years, which was within the range given in the definition of EOAD6. The literature review in this regard suggested a variation in the mean age of EOAD even within the same population. For an instance, the mean age in one Dutch study was 60 ± 4 years10. In another Dutch study, the mean age was 56 ± 5 years11. This variation in our opinion, depended on the period after which an established diagnosis of EOAD was given. We usually expect a relatively later onset for the disease in developing countries due to the lack of modern diagnostic tools. However, the mean age for EOAD in our study did not vary much with literature in this context.
Our findings regarding gender suggested that females were more affected than males (72.4% vs. 27.6%). Our findings agreed with the general assumption that Alzheimer’s disease occurs primarily in women as suggested by Mary12 , despite the fact that some studies did not discriminate between males and females12. In her study, Mary has attributed this variation in gender to hormonal and psychological factors and pregnancy-related morbidities12. It was established that the female hormones oestrogen and progesterone increased the risk for dementia12. Mental disorders related to pregnancy such as psychosis and post-partum depression were also known to aggravate the cognitive impairment in AD12.
When we looked into the mean age and gender together, we could see that our patients were mainly post-menopausal women. Therefore, our results supported the study by Geeske13 who suggested that oestrogen may protect against neuronal degeneration, and hence postmenopausal women have a greater risk of AD than perimenopausal women or women taking hormone replacement therapy. From our point of view, the lack of oestrogen in our patients might have played a role in the acceleration of the neuronal degeneration seen in AD patients. Our findings confirmed that age and gender have altered the correlation between MTHFR C677T polymorphism and EOAD to some degree.
The frequency of MTHFR C677T in the current study was 39.7% for the heterozygous allele and 57.5% for the homozygous allele. Both frequencies were within the proposed range for the t alle prevalence of 12-57%28. The current study concluded that both the homozygous (TT) and the heterozygous alleles (CT) of MTHFR C677T polymorphism were associated with EOAD in Sudanese population. Because the mutation was associated with the early onset disease, our findings supported the conclusion drawn by Yaling28 that MTHFR C677T polymorphism might advance the onset of the disease. The current study, however, did not find a significant association between the disease and A1298C polymorphism, supporting the fact that MTHFR C677T is the most clinically significant polymorphism associated with AD among the three polymorphisms28. Our study, however, could not finalize whether this polymorphism might be considered as a risk factor for AD or not29,30. Our results also confirmed the findings of the study by Ye Hu31 who concluded that both the heterozygous and homozygous alleles of MTHFR C677T were associated with AD.
Considering A1298C polymorphisms, we could not totally agree with the case study by Leila32 who detected both C677T and A1298C polymorphisms in a patient with EOAD32. Furthermore, we could not support or deny whether A1298C polymorphism has a causative33, or a protective28 role in the pathogenesis of AD28. Because EOAD is basically multifactorial, we could not conclude that C677T polymorphism was the sole etiology behind AD4.
The current study has confirmed a significant elevation in plasma homocysteine levels among cases, suggesting a link between MTHFR 677T mutation and AD. It was already proven that MTHFR C677T polymorphism causes hyperhomocysteinemia41, which is associated with AD53. Furthermore, it was already established that elevated homocysteine levels might induce white matter signal abnormalities34 that can be responsible for the impaired cognitive function in patients with AD34. This assumption, however, was not conclusive because of the multifactorial nature of AD and the fact that both genetic and environmental factors together decide who will get the disease4. The low folate probably due to MTHFR C677T polymorphism was thought to ameliorate the cognitive impairment seen in patients with AD36. It was suggested that hyperhomocysteinemia might be caused by factors other than MTHFR C677T polymorphism. These factors included low folate and vitamin B12 levels, aging, smoking and renal impairment34. Because we did not measure folate and B12 levels in our participants, we could not confirm whether low folate level has contributed to the cognitive impairment or not. Moreover, we could not exclude low folate as a cause of hyperhomocysteinemia. Interestingly, our findings clashed with one study by Luchsinger40 who found no significant association between high plasma homocysteine levels and AD. We concluded that individuals with elevated plasma homocysteine levels were more likely to develop EOAD than individuals with normal homocysteine levels.
We have also looked into the means of plasma homocysteine levels within patients having CC, CT, and TT genotypes. Our results suggested that homocysteine levels were significantly elevated in the group with the homozygous allele, followed by the heterozygous allele, whereas levels were within the normal range in the group having the wildtype allele. This finding supported the conclusion drawn by Munshi38 who reported higher plasma homocysteine levels in the TT genotype, followed by the CT then the CC genotype.
Conclusion
In conclusion, the frequency of EOAD among Sudanese population was as low as 0.0012%. The mean age of EOAD patients was 57.9 ±3 years. Females were more commonly affected than males due to hormonal and psychological variation. The frequency of MTHFR C677T in the current study was 39.7% for the heterozygous allele and 57.5% for the homozygous allele. Both the homozygous and heterozygous alleles were associated with EOAD. It appears that the mutation might advance the onset of the disease and the elevated plasma homocysteine levels might be partially responsible for the neurodegenerative abnormalities seen in those patients. Our results suggested that homocysteine levels were significantly increased in the group with the homozygous allele, followed by the heterozygous allele, whereas levels were within the normal range in the group having the wildtype allele. MTHFR A1298C polymorphism was not associated with EOAD in the study. Individuals with family history of Alzheimer’s disease may need to be screened for this mutation as early as possible, so that proper preventive and therapeutic measures could be implemented once the polymorphism is detected. MTHFR C677T-positive individuals should be considered at-risk for progression to Alzheimer’s disease.
The study was without limitations. We could not exclude all factors that might elevate plasma homocysteine levels like diet, low folate and B12 levels. Additionally, we were unable to establish a direct effect or causation as a case-control study. Moreover, the study might be less useful for examining the risk factors for such a rare disease as in cohort studies.
Acknowledgement
The authors would like to thank the staff at Khartoum Teaching Hospital, Al-Shaab Teaching Hospital, Al-Tijani Al-Mahi Neuropsychiatric Hospital and the Research Centre at the Faculty of Medicine, El-Neelain University for their kind help in recruiting study subjects and collecting samples.
Funding Sources
The author(s) received no financial support for the research, authorship, and/or publication of this article
Conflict of Interest
The authors do not have any conflict of interest
References
- Soria Lopez JA, González HM, Léger GC. Alzheimer’s disease. Handbook of Clinical Neurology. 2019;167:231-255. doi: https://doi.org/10.1016/b978-0-12-804766-8.00013-3.
CrossRef - Kumar A, Tsao JW, Sidhu J, Goyal A. Alzheimer Disease. Nih.gov. Published June 5, 2022. https://www.ncbi.nlm.nih. gov/books/NBK499922/
CrossRef - Dumurgier J, Sabia S. [Epidemiology of Alzheimer’s disease: latest trends]. La Revue Du Praticien. 2020;70(2):149-151. https://pubmed.ncbi.nlm.nih.gov/32877124/
CrossRef - Zhang XX ., Tian Y, Wang ZT ., Ma YH ., Tan L, Yu JT . The Epidemiology of Alzheimer’s Disease Modifiable Risk Factors and Prevention. The Journal of Prevention of Alzheimer’s Disease. 2021;8(8):1-9. doi: https://doi.org/10.14283/jpad.2021.15.
CrossRef - Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues in clinical neuroscience. 2009;11(2):111-128.
CrossRef - Mendez MF. Early-Onset Alzheimer Disease. Neurologic Clinics. 2017;35(2):263-281. doi: https://doi.org/10.1016/j.ncl.2017.01.005.
CrossRef - Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL. Alzheimer’s disease. Nature Reviews Disease Primers. 2015;1(15056):15056. doi: https://doi.org/10.1038/nrdp.2015.56.
CrossRef - Qin Q, Yin Y, Wang Y, Lu Y, Tang Y, Jia J. Gene mutations associated with early onset familial Alzheimer’s disease in China: An overview and current status. Molecular Genetics & Genomic Medicine. 2020;8(10). doi: https://doi.org/10.1002/mgg3.1443.
CrossRef - Sheppard O, Coleman M. Alzheimer’s Disease: Etiology, Neuropathology and Pathogenesis. PubMed. Published 2020. https://pubmed.ncbi.nlm.nih.gov/33400468/
- Smits LL, Pijnenburg YAL, Koedam ELGE, et al. Early Onset Alzheimer’s Disease is Associated with a Distinct Neuropsychological Profile. Journal of Alzheimer’s Disease. 2012;30(1):101-108. doi: https://doi.org/10.3233/jad-2012-111934.
CrossRef - Koedam ELGE, Lauffer V, van der Vlies AE, van der Flier WM, Scheltens P, Pijnenburg YAL. Early-Versus Late-Onset Alzheimer’s Disease: More than Age Alone. Journal of Alzheimer’s Disease. 2010;19(4):1401-1408. doi: https://doi.org/10.3233/jad-2010-1337.
CrossRef - O’Neal MA. Women and the risk of Alzheimer’s disease. Frontiers in global women’s health. 2024;4. doi: https://doi.org/10.3389/ fgwh.2023.1324522.
CrossRef - 1.Peeters G, Katelekha K, Lawlor B, Demnitz N. Sex differences in the incidence and prevalence of young‐onset Alzheimer’s disease: A meta‐analysis. International Journal of Geriatric Psychiatry. 2021;37(1). doi: https://doi.org/10.1002/gps.5612.
CrossRef - Ayodele T, Rogaeva E, Kurup JT, Beecham G, Reitz C. Early-Onset Alzheimer’s Disease: What Is Missing in Research? Current Neurology and Neuroscience Reports. 2021;21(2). doi: https://doi.org/10.1007/ s11910-020-01090-y.
CrossRef - Frota NAF, Nitrini R, Damasceno BP, et al. Criteria for the diagnosis of Alzheimer’s disease: Recommendations of the Scientific Department of Cognitive Neurology and Aging of the Brazilian Academy of Neurology. Dementia & Neuropsychologia. 2011;5(3):146-152. doi: https://doi.org/10.1590/s1980-57642011dn05030002.
CrossRef - Zhang C. Etiology of Alzheimer’s Disease. Discovery Medicine. 2023;35(178):757-757. doi: https://doi.org/10.24976/ discov.med.202335178.71
CrossRef - Breijyeh Z, Karaman R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules. 2020;25(24):5789. doi: https://doi.org/10.3390/molecules25245789.
CrossRef - A. Armstrong R. Risk Factors for Alzheimer’s Disease. Folia Neuropathologica. 2019;57(2):87-105. doi: https://doi.org/10.5114/ fn.2019.85929
CrossRef - Abubakar MB, Sanusi KO, Ugusman A, et al. Alzheimer’s Disease: An Update and Insights Into Pathophysiology. Frontiers in Aging Neuroscience. 2022;14(1):742408. doi: https://doi.org/10.3389/ fnagi.2022.742408.
CrossRef - Goel P, Chakrabarti S, Goel K, Bhutani K, Chopra T, Bali S. Neuronal Cell Death Mechanisms in Alzheimer’s disease: an Insight. Frontiers in Molecular Neuroscience. 2022;15. doi: https://doi.org/10.3389/ fnmol.2022.937133.
CrossRef - Planche V, Bouteloup V, Mangin J, et al. Clinical relevance of brain atrophy subtypes categorization in memory clinics. Alzheimer’s & Dementia. 2020;17(4):641-652. doi: https://doi.org/10.1002/alz.12231.
CrossRef - Sochocka M, Zwolińska K, Leszek J. The Infectious Etiology of Alzheimer’s Disease. Current Neuropharmacology. 2017;15(7). doi: https://doi.org/10.2174/1570159×15666170313122937.
CrossRef - Cummings JL, Vinters HV, Cole GM, Khachaturian ZS. Alzheimer’s disease: Etiologies, pathophysiology, cognitive reserve, and treatment opportunities. Neurology. 1998;51(Issue 1, Supplement 1):S2-S17. doi: https://doi.org/10.1212/wnl.51.1_suppl_1.s2
CrossRef - Yarns BC, Holiday KA, Carlson DM, Cosgrove CK, Melrose RJ. Pathophysiology of Alzheimer’s Disease. The Psychiatric Clinics of North America. 2022;45(4):663-676. doi: https://doi.org/10.1016/ j.psc.2022.07.003.
CrossRef - Jiang T, Yu JT, Tian Y, Tan L. Epidemiology and Etiology of Alzheimer’s disease: From Genetic to Non- Genetic Factors. Current Alzheimer Research. 2013;10(8):852-867. doi: https://doi.org/10.2174/ 15672050113109990155.
CrossRef - Hoogmartens J, Cacace R, Van Broeckhoven C. Insight into the genetic etiology of Alzheimer’s disease: A comprehensive review of the role of rare variants. Alzheimer’s & Dementia: Diagnosis, Assessment & Disease Monitoring. 2021;13(1). doi: https://doi.org/10.1002/dad2.12155.
CrossRef - Lacour M, Quenez O, Rovelet-Lecrux A, et al. Causative Mutations and Genetic Risk Factors in Sporadic Early Onset Alzheimer’s Disease Before 51 Years. Journal of Alzheimer’s Disease. 2019;71(1):227-243. doi: https://doi.org/10.3233/JAD-190193.
CrossRef - Jiang Y, Xiao X, Wen Y, et al. Genetic effect of MTHFR C677T, A1298C, and A1793G polymorphisms on the age at onset, plasma homocysteine, and white matter lesions in Alzheimer’s disease in the Chinese population. Aging. 2021;13(8):11352-11362. doi: https://doi.org/10.18632/aging.202827.
CrossRef - Peng Q, Lao X, Huang X, Qin X, Li S, Zeng Z. The MTHFR C677T polymorphism contributes to increased risk of Alzheimer’s Disease: Evidence based on 40 case-control studies. Neuroscience Letters. 2015;586:36-42. doi: https://doi.org/10.1016/j.neulet.2014.11.049
CrossRef - Yi J, Xiao L, Zhou SQ, Zhang WJ, Liu BY. The C677T polymorphism of the methylenetetrahydrofolate reductase gene and susceptibility to late-onset Alzheimer’s disease. Open Medicine. 2019;14(1):32-40. doi: https://doi.org/10.1515/med-2019-0006.
CrossRef - Hua Y, Zhao H, Kong Y, Ye M. Association Between the MTHFR Gene and Alzheimer’s Disease: A Meta-Analysis. Int J Neurosci. 2011;121 (8):462-471. doi: https://doi.org/10.3109/00207454.2011.578778
CrossRef - Mansouri L, Klai S, Fekih-Mrissa N, Gritli N, Mrissa R. C677T and A1298C gene polymorphisms and sporadic early-onset Alzheimer’s disease. Advances in Alzheimer’s Disease. 2013;02(04):132-134. doi: https://doi.org/10.4236/aad.2013.24018.
CrossRef - Mansouri L, Fekih-Mrissa N, Klai S, Mansour M, Gritli N, Mrissa R. Association of methylenetetrahydrofolate reductase polymorphisms with susceptibility to Alzheimer’s disease. Clinical Neurology and Neurosurgery. 2013;115(9):1693-1696. doi: https://doi.org/10.1016/ j.clineuro.2013.03.015.
CrossRef - Hainsworth AH, Yeo NE, Weekman EM, Wilcock DM. Homocysteine, hyperhomocysteinemia and vascular contributions to cognitive impairment and dementia (VCID). Biochimica et Biophysica Acta (BBA) – Molecular Basis of Disease. 2016;1862(5):1008-1017. doi: https://doi.org/10.1016/j.bbadis.2015.11.015.
CrossRef - Hoffman A, Taleski G, Qian H, et al. Methylenetetrahydrofolate Reductase Deficiency Deregulates Regional Brain Amyloid-β Protein Precursor Expression and Phosphorylation Levels. Götz J, ed. Journal of Alzheimer’s Disease. 2018;64(1):223-237. doi: https://doi.org/10.3233/jad-180032.
CrossRef - Ma F, Wu T, Zhao J, et al. Plasma Homocysteine and Serum Folate and Vitamin B12 Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Case-Control Study. Nutrients. 2017;9(7). doi: https://doi.org/10.3390/nu9070725.
CrossRef - Raghubeer S, Matsha TE. Methylenetetrahydrofolate (MTHFR), the One-Carbon Cycle, and Cardiovascular Risks. Nutrients. 2021;13(12):4562. doi: https://doi.org/10.3390/nu13124562.
CrossRef - Munshi R, Panchal F, Kulkarni V, Chaurasia A. Methylenetetrahydrofolate reductase polymorphism in healthy volunteers and its correlation with homocysteine levels in patients with thrombosis. Indian journal of pharmacology. 2019;51(4):248-254. doi: https://doi.org/10.4103/ijp.IJP_215_19.
CrossRef - Adelekan OO, Uche EI, Balogun TM, et al. Methylene tetrahydrofolate reductase gene mutation in sickle cell anaemia patients in Lagos, Nigeria. Pan African Medical Journal. 2019;34. doi: https://doi.org/10.11604/pamj.2019.34.213.19524.
CrossRef - Luchsinger JA, Tang MX ., Shea S, Miller J, Green R, Mayeux R. Plasma homocysteine levels and risk of Alzheimer disease. Neurology. 2004;62(11):1972-1976. doi: https://doi.org/10.1212/01.wnl. 0000129504.60409.88.
CrossRef - Froese DS, Huemer M, Suormala T, et al. Mutation Update and Review of Severe Methylenetetrahydrofolate Reductase Deficiency. Human Mutation. 2016;37(5):427-438. doi: https://doi.org/10.1002/humu.22970.
CrossRef - Zhang X, Hou C, Liu P, et al. Methylenetetrahydrofolate Reductase (MTHFR) C677T Polymorphism and Subacute Combined Degeneration: Revealing a Genetic Predisposition. Frontiers in Neurology. 2019;9. doi: https://doi.org/10.3389/fneur.2018.01162.
CrossRef - Hamad NA, Eltayeb LB. New Insight of Methylenetetrahydrofolate Reductase (MTHFR) C677T Gene Polymorphisms, and Serum Electrolytes in Cardiac Syndrome X Patients. Journal of Pharmaceutical Research International. Published online December 7, 2021:115-121. doi: https://doi.org/10.9734/jpri/2021/v33i53b33687.
CrossRef - Esposito A, Monda E, Gragnano F, et al. Prevalence and clinical implications of hyperhomocysteinaemia in patients with hypertrophic cardiomyopathy and MTHFR C6777T polymorphism. European Journal of Preventive Cardiology. 2020;27(17):1906-1908. doi: https://doi.org/10.1177/2047487319888596
CrossRef - Rafik A, Rachad L, Kone A, Nadifi S. MTHFR C677T polymorphism and risk of nonsyndromic cleft lip with or without cleft palate in the Moroccan population. The Application of Clinical Genetics. 2019;Volume 12:51-54. doi: https://doi.org/10.2147/tacg.s194166.
CrossRef - Nigam N, Singh PK, Agrawal M, Nigam S, Gupta H, Saxena S. MTHFR C677T, Prothrombin G20210A, and Factor V Leiden (G1691A) Polymorphism and Beta-Thalassemia Risk: A Meta-Analysis. Cureus. Published online September 30, 2020. doi: https://doi.org/10.7759/cureus.10743.
CrossRef - Azimi-Nezhad M, Teymoori A, Salmaninejad A, Ebrahimzadeh-Vesal R. Association of MTHFR C677T Polymorphism with Preeclampsia in North East of Iran (Khorasan Province). Fetal and Pediatric Pathology. 2019;39(5):373-380. doi: https://doi.org/10.1080/15513815.2019.1655819.
CrossRef - Elqadi M, Eweidat K, Abu Sabha M, et al. Methylenetetrahydrofolate reductase C677T gene polymorphism and the association with dyslipidemia in type 2 diabetic Palestinian patients. Journal of Clinical Laboratory Analysis. 2021;35(10). doi: https://doi.org/10.1002/ jcla.23994.
CrossRef - Stanojevic A, Jelena Spasic, Marinkovic M, et al. Methylenetetrahydrofolate reductase polymorphic variants C677T and A1298C in rectal cancer in Slavic population: significance for cancer risk and response to chemoradiotherapy. Frontiers in genetics. 2024;14. doi: https://doi.org/10.3389/fgene.2023.1299599.
CrossRef - Rutten-Jacobs LCA, Traylor M, Adib-Samii P, et al. Association of MTHFR C677T Genotype With Ischemic Stroke Is Confined to Cerebral Small Vessel Disease Subtype. Stroke. 2016;47(3):646-651. doi: https://doi.org/10.1161/strokeaha.115.011545.
CrossRef - Tabatabaei RS, Fatahi-Meibodi N, Meibodi B, et al. Association of Fetal MTHFR C677T Polymorphism with Susceptibility to Neural Tube Defects: A Systematic Review and Update Meta-Analysis. Fetal and Pediatric Pathology. Published online June 14, 2020:1-17. doi: https://doi.org/10.1080/15513815.2020.1775734.
CrossRef - Wu D, Shi D, Yang L, Zhu X. Association between methylenetetrahydrofolate reductase C677T polymorphism and psoriasis: A meta-analysis. The Journal of Dermatology. 2015;43(2):162-169. doi: https://doi.org/10.1111/1346-8138.13039.
CrossRef - Stoccoro A, Tannorella P, Salluzzo MG, et al. The Methylenetetrahydrofolate Reductase C677T Polymorphism and Risk for Late-Onset Alzheimer’s disease: Further Evidence in an Italian Multicenter Study. Journal of Alzheimer’s Disease. 2017;56(4):1451-1457. doi: https://doi.org/10.3233/jad-161081.
CrossRef - Zhang YX, Yang LP, Gai C, et al. Association between variants of MTHFR genes and psychiatric disorders: A meta-analysis. Frontiers in Psychiatry. 2022;13. doi: https://doi.org/10.3389/fpsyt.2022.976428.
CrossRef - Graydon JS, Claudio K, Baker S, et al. Ethnogeographic prevalence and implications of the 677C>T and 1298A>C MTHFR polymorphisms in US primary care populations. Biomarkers in Medicine. 2019;13(8):649-661. doi: https://doi.org/10.2217/bmm-2018-0392.
CrossRef - Long S, Goldblatt J. MTHFR genetic testing: Controversy and clinical implications. Australian Family Physician. 2016;45(4):237-240. https://pubmed.ncbi.nlm.nih.gov/27052143/
- Althubaiti A. Sample Size determination: a Practical Guide for Health Researchers. Journal of General and Family Medicine. 2022;24(2):72-78. doi: https://doi.org/10.1002/jgf2.600.
CrossRef - Rodríguez del Águila M, González-Ramírez A. Sample size calculation. Allergologia et Immunopathologia. 2014;42(5):485-492. doi: https://doi.org/10.1016/j.aller.2013.03.008.
CrossRef - Chen WC, Kerr R, May A, et al. The Integrity and Yield of Genomic DNA Isolated from Whole Blood Following Long-Term Storage at −30°C. Biopreservation and Biobanking. 2018;16(2):106-113. doi: https://doi.org/10.1089/bio.2017.0050.
CrossRef - Ludeman MJ, Zhong C, Mulero JJ, et al. Developmental validation of GlobalFilerTM PCR amplification kit: a 6-dye multiplex assay designed for amplification of casework samples. International Journal of Legal Medicine. 2018;132(6):1555-1573. doi: https://doi.org/10.1007/s00414-018-1817-5
CrossRef - Bruzzese L, Fenouillet E, Julien Fromonot, et al. High homocysteine levels prevent via H2S the CoCl2‐induced alteration of lymphocyte viability. Journal of Cellular and Molecular Medicine. 2016;20(8):1411-1419. doi: https://doi.org/10.1111/jcmm.12829.
CrossRef - Sui X, Yang Z, Wang F, et al. The relationship between plasma homocysteine levels and MTHFR gene variation, age, and sex in Northeast China. Nigerian Journal of Clinical Practice. 2019;22(3):380-380. doi: https://doi.org/10.4103/njcp.njcp_291_18.
CrossRef

