
Manuscript accepted on :25-03-2022
Published online on: 30-03-2022
Plagiarism Check: Yes
Reviewed by: Dr. Subhasis Chakraborty
Second Review by: Dr. Ajoy Kumer
Final Approval by: Dr. Ian james martin
Christina Nilofer and Arumugam Mohanapriya*
and Arumugam Mohanapriya*
School of Biosciences and Technology, VIT University, Vellore, Tamil Nadu, India
Corresponding Author E-mail:mohanapriyaa@vit.ac.in
DOI : https://dx.doi.org/10.13005/bpj/2383
Abstract
The coronavirus outbreak and its mutant variants have harmed the health of the human populace and imperiled the world economy. Several studies are initiated across the globe using clinical biomarkers from hematological, immunological, and biochemical experiments. In addition, analysis of protein interfaces provides an understanding of the functioning of the coronavirus target proteins. This study examines the interfaces of spike glycoproteins in terms of large (vdW dominant) and small (vdW subdominant) interfaces. We also calculated Gibbs free energy (ΔG), residue propensity and hot-spot prediction for these interfaces. Dataset consisting of 115 (large interface with vdW dominant) and 18 (small interface with vdW subdominant) were obtained from PDB. Results show that 86% of the total interfaces were vdW dominant, while the rest, 14%, were sub-dominant in vdW energy. Interestingly, on average, we found the Gibbs free energy (ΔG) of large and small interfaces to be -21 and -30 kcal/mol respectively. We also found the interfaces of large and small to be highly pronounced with polar residues followed by hydrophobic residues in case of large interfaces and charged residues in case of small interfaces. We found and report methionine residues to be absent at the small interfaces having subdominant vdW energy. We also observed the majority of the interfaces to be rich in hotspot residues. Thus, the information on heteromeric interactions of glycoproteins may help develop new and productive therapeutic drugs.
Keywords
Area; Coronavirus; Electrostatic; Heteromers; H-Bonds; Hotspot; Residue Propensity; Spike glycoproteins
Download this article as:| Copy the following to cite this article: Nilofer C, Mohanapriya A. Electrostatic Interactions Contribute to the Overall Structural Stability in Small Interfaces of Corona Viral Spike Glycoproteins. Biomed Pharmacol J 2022;15(1). |
| Copy the following to cite this URL: Nilofer C, Mohanapriya A. Electrostatic Interactions Contribute to the Overall Structural Stability in Small Interfaces of Corona Viral Spike Glycoproteins. Biomed Pharmacol J 2022;15(1). Available from: https://bit.ly/3x4mpUf |
Introduction
The impact of the COVID-19 pandemic on the world’s economy and overall health of the human populace has been devastating, with mortalities amounting to over 5 million and cases still raising, and countries suffering crumbling hits to their GDPs. The repercussions of this pandemic leave room for no doubt about the need to characterize spike glycoproteins, the etiological agent of COVID-19. The urgency of the situation becomes all the more alarming with the rise of the various SARS-CoV-2 variants, each one being a reminder of the looming threat of the virus’s capability to mutate further and further to evade and overcome the vaccines being developed and deployed against it (https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/, https://covid19.who.int/). Mutations may be a cause for concern in humans. Still, in viruses, it’s a matter of survival, a chance at becoming more infectious and pathogenic, and evading its hosts’ immune system. Since its advent, many variants of SARS CoV-2 have stemmed from it. Of the many that exist, four, namely the alpha, the beta, the gamma, and the delta, pose a severe threat to human health and welfare (https://www.livemint.com/science/health/from-alpha-beta-gamma-to-delta-what-we-know-of-coronavirus-mutations-so-far-11625207686643.html).
Many efforts worldwide have made it possible to gain a comprehensive view of the viral architecture of coronavirus spike glycoprotein, furthering our understanding of the virus’s modus operandi and enabling us to leverage that knowledge in devising effective and efficient actions against COVID. Ye et al., 2020 stated that a significant amount of measures have been taken in this hunt of eradicating COVID-19 from the beginning of the pandemic. A varied range of solutions has been raised from the persistent efforts of many scientists and researchers across the globe (Odolczyk et al., 2021; Auwaerter and Casadevall, 2020; Yang, 2021; Ahsan et al., 2020). Research on the functioning of SARS-CoV-2 has provided us with new particulars about the linchpins and its viral constitution, as they serve as a potential drug targets (Sakkiah et al., 2021; Xie et al., 2020; Gordon et al., 2020). Regardless of their host specificities, all coronaviruses have some common proteins and are crucial to their life cycle (Chang et al., 2021). Amongst such proteins, the spike protein has received particular interest. Through its receptor-binding domain, the spike glycoprotein recognizes and attaches to its cognate receptor on the host cells. This binding expedites the virus’s cellular entry. Thus, its presence and functioning are vital to kickstart the virus’s life cycle, and having an idea about the interface interactions and the Gibbs free energies of the spike glycoproteins and all its associated variants will be most suitable if we are to win this war against this virus.
A critical aspect of coming up with a beneficial remedy is by assessing the interactions that occur at the protein interface of the essential proteins, pivotal to the organism’s pathogenicity. Proteins are the sine qua non of almost all biological processes. Two or more proteins come together in a biological system to perform a molecular function including signal transduction and cellular transport through the formation of a stable interface (Hardcastle et al. 2017). Protein complexes have shown its capabilities in therapeutics as it’s increasingly received a lot of attention in the past few years. Chang et al. 2021 states that the protein interface would be the ideal targets for designing antiviral drugs as it enriches the targetable chemical space by providing alternative targets for drug discovery. Therefore, any perturbations in their interface interactions result in irregularities in their structure and function, by either decreasing their effectiveness or becoming non-functional. Consequently, it is crucial to obtain meaningful insights from the interfaces of spike glycoproteins through characterization as well to acquire the intellectual wherewithal to beat the menace of coronavirus and the various variants that have arisen in its wake.
Since 1975, protein interfaces are been characterized using several physicochemical features including interface area and size (Chothia & Janin, 1975; Chothia et al., 1976; Xu et al., 1997; Porter et al., 2019; Dauzhenka et al., 2018;), hydrogen bonds (Tsai et al., 2008; Bahadur et al., 2004; Li et al., 2006; Lo Conte et al., 1999; Miller et al., 1987;), shape complementarity (Caffrey et al., 2004; Correa Marrero et al., 2019; Dai et al., 2016;), gap volume index (Jones & Thornton, 1995, 1996, 1997a, 1997b;), hydration (Korn & Burnett, 1991; Robert & Janin, 1998;), vdW energy force (Nilofer et al., 2020;), hydrophobicity, hydrophilicity (Murakami & Jones, 2006; Zhanhua et al., 2005;), aromatic residues (Gromiha et al., 2009; Li et al., 2019;), residue propensity (Pal et al., 2007; Chakrabarti & Janin, 2002; Guharoy & Chakrabarti, 2010; Guharoy & Sowmya et al., 2011;), electrostatics energy (Nilofer et al., 2017;), preserved residues (Janin & Chothia, 1990;), configuration and conformational changes (Hwang et al., 2016; Humphris & Kortemme, 2008;Sowmya et al., 2015; Sowmya & Ranganathan, 2015; Marchetti et al., 2019;), Gibbs free binding energy (Guo et al., 2016; Daberdaku & Ferrari, 2018; Yang & Gong, 2018;) and the presence of water molecules at the interface (Taechalertpaisarn et al., 2019; Garcia-Garcia et al., 2017; Li & Kihara, 2012; Bendell et al., 2014; Jordan et al., 2012; Qiao et al., 2018; Moreira et al., 2017; Xue et al., 2011; Wang et al., 2017;) These characterizations of protein interfaces have intensified our understanding about the function mechanism of the two interacting proteins. Our previous study states that the small interfaces of the two interacting proteins have small Interface Size and Interface Area are rich in electrostatic energy (Nilofer et al 2017 & 2020). Therefore, it is of interest to examine and compare the large (vdW dominant) and small (vdW subdominant) interfaces of heteromeric coronaviral spike glycoproteins using Interface Area, Interface Size, vdW, Electrostatic, Hydrogen bond and total energies, Gibbs free energy (binding energy), residue propensity (calculated using an in-house python code) and hotspot prediction.
Materials and Methods
Dataset
A structural dataset (non-redundant and updated) of 133 heteromeric coronaviral spike glycoproteins were obtained from PDB. The dataset were created by collecting protein structures that were found to satisfy the following conditions: Method: X-Ray Diffraction, Type: proteins, Refinement Resolution: less than 3.0 Å, and Sequence Length: greater than 50 aa. Furthermore, the obtained 133 protein complexes were classified into large and small interfaces based on their Interface size, Interface Area and vdW contribution at the protein interface. Hence 133 interfaces gave rise to 115 large interfaces with vdW dominant and 18 small interfaces having subdominant vdW. These interfaces were analyzed and compared using protein’s Interface size, Interface Area, vdW, Electrostatic, Hydrogen bond and total binding energy, Gibbs free energy (ΔG), residue propensity and hotspots.
Interface Size
Interface Size refers to the number of amino acid residues at the binding region of the two interacting proteins.
Interface Area
Interface Area for all the large and small interfaces were calculated using NACCESS (Hubbard, 1993) using the principle: [(Accessible Surface Area (ASA) of Subunit A + ASA of Subunit B) – (ASA of the dimer (AandB))] / 2. ASA of a molecule is calculated using atomic coordinate information of its PDB file. It works on the Lee and Richard’s principle (Lee & Richards, 1971), wherein a probe of 1.4Å radius (Jones & Thornton, 1996) is rolled over the surface of a protein in a bound and unbound state to compute the ASA. 1.4Å is used as a probe radius in ASA calculations as it is the water molecule’s radius.
Interface Energy
Interface energy refers to the total amount of energy contributed by vdW, hydrogen bonds, and electrostatic energies summing up to total stabilizing energy at the protein interface are referred to as interface energies. These interface energies were calculated for all of the large and small protein interfaces. The analysis was done with the aid of PPCheck (public web-server), it is helpful in computing the non-covalent interaction energies of a protein complex using their atomic coordinate information (Sukhwal & Sowdhamini, 2013 and 2015). It should be noted that the role of interface water was expelled in the estimation.
Large and small interfaces
Protein interfaces with interface area less than 1000 Å are grouped as small interfaces and interfaces with interface area greater than 1000 Å are grouped as large interfaces.
Dominant and subdominant vdW interfaces
An interface with vdW contribution >60 % were defined as vdW dominant and rest as vdW sub-dominant. A cutoff of 60% was chosen because most of the interfaces were with vdW energy at 60% cutoff.
Gibbs free energy (ΔG)
Gibbs free energy for each of small and large protein interfaces were calculated using PDBePISA online web server (Krissinel and Henrick, 2007). PDBePISa calculates the Gibbs free energy of a protein interface both in bound and unbound state. The Gibbs free energy for large and small interfaces of coronoviral heteromers are provided in the supplementary excel sheet.
Residue propensity
Residue propensity refers to the frequency of occurrence of a particular amino acid residue at the interface of two interacting proteins. Prior to residue propensity calculation, the interface residues were computed using the InterProSurf server (Negi et al, 2007). InterProSurf is an online software build on Perl modules for calculating the amino acid residues found at the interface of two interacting proteins. Subsequently, residue propensities were calculated using the in-house python code (Supplementary material 1).
Hotspot prediction
Residues possessing high energy residues with normalized energy are defined as Hotspot residues. These Hotspot residues were computed using the PPCheck server (Sukhwal & Sowdhamini, 2013, 2015). PPCheck computes the degree of interaction and normalized energies for each interface residue.
Statistical analysis
Statistical parameters like mean, standard deviation and multiple linear regressions were calculated for the large and small protein interfaces using Interface size, area and energy with the statistical tools (regression) in MS Office Excel 2007. Finally, the coefficient of determination (r2), an analytical power score, was determined with an evaluation of significance (p-value) using a statistical ANOVA test at a 95% confidence limit. The evaluation was considered significant when p < 0.01.
Results
The primary aim of this study is to characterize and compare the large and small heteromeric interfaces of spike glycoproteins using non-covalent interactions that stabilize the interface. Only when we know what keeps a system together can we work towards destabilizing it. PPcheck (a distance-based, non-covalent interaction quantifying webserver) proved to be of immense help in gauging the stabilizing energies of the protein-protein interfaces. Interface attributes like interface size, interface area, van der Waals, Hydrogen bonds, electrostatic, and total energy were estimated for the large and small heteromeric interfaces along with the prediction of Gibbs free energy (ΔG), residue propensity and hotspot prediction, the dataset for which is presented in Table 1.
Table 1: List of heteromeric coronaviral spike glycoproteins with large and small interfaces having dominant and subdominant vdW (van der Waals energy).
| Heteromeric coronoviral spike glycoproteins – large interface with vdW dominant (115) | ||||||||
| 2dd8 | 5yy5 | 6yla | 7bz5 | 7d30 | 7jmp | 7kmi | 7m53 | 7nx9 |
| 2ghw | 6c6z | 6yz5 | 7c01 | 7dc6 | 7jmw | 7kn5 | 7m55 | 7nxa |
| 3d0i | 6j2j | 6z2m | 7c8v | 7deo | 7jn5 | 7kn6 | 7m7w | 7nxb |
| 4kr0 | 6m0j | 6zcz | 7c8w | 7det | 7jx3 | 7kn7 | 7m8s | |
| 4qzv | 6pxh | 7b3o | 7can | 7deu | 7k9z | 7kzb | 7m8t | |
| 4xak | 6u7f | 7beh | 7cdi | 7djz | 7kfv | 7l0n | 7m8u | |
| 4zpt | 6vw1 | 7bei | 7chb | 7e7x | 7kfw | 7ldj | 7mf1 | |
| 5do2 | 6waq | 7bej | 7chc | 7e7y | 7kfx | 7lm8 | 7mfu | |
| 5gmq | 6xc3 | 7bek | 7chf | 7e86 | 7kfy | 7lm9 | 7mmo | |
| 5gr7 | 6xc4 | 7bel | 7cho | 7e8m | 7kgj | 7lo4 | 7neg | |
| 5gsb | 6xc7 | 7bem | 7chp | 7eam | 7kgk | 7lop | 7neh | |
| 5gsr | 6xe1 | 7ben | 7chs | 7ean | 7klw | 7m3i | 7nx6 | |
| 5gsv | 6xkp | 7bep | 7cjf | 7jjc | 7kmg | 7m51 | 7nx7 | |
| 5gsx | 6xkq | 7bwj | 7d2z | 7jmo | 7kmh | 7m52 | 7nx8 | |
| Heteromeric coronoviral spike glycoproteins – small interface with vdW subdominant (18) | ||||||||
| 4zpt | 6u7f | 6xc3 | 6z2m | 7bep | 7jjc | 7kfw | 7l0n | 7m7w |
| 6c6z | 6u7g | 6yla | 7bel | 7deo | 7jn5 | 7kmh | 7m52 | 7mmo |
Furthermore, various statistical analyses were performed on the obtained data to advance our comprehension of the workings of the heteromeric interfaces of spike glycoprotein. For any protein-protein interface, the strength of the interactions is governed by the number of residues that constitute the interface and the area across which the interactions come into play says Chothia et al, 1975. Logically, an increase in the interface size should entail an increase in the area of a protein-protein interface. Thus, both structural properties should exhibit a positive correlation. It is a well-known fact the interface size increases with the interface area (Chothia et al, 1996 & Jones et al, 1995). The average interface size and interface area of the total interfaces are 57 ± 25 and 118 ± 72, respectively. However, the interface area and interface size of large (120 ± 73 and 58 ± 26) and small (93 ± 61) (43 ± 19) glycoprotein’s differs accountably. We found 86% of the total interfaces to be large interface with large interface area and interface size whereas, rest 14% to be small interface with small interface area and interface size.
 |
Figure 1: Illustration of an example large and small heteromeric interface of a coronavirus spike glycoprotein (PDB ID: 2DD8). |
The large and small heteromeric interfaces were described using interface area, interface size, and interface energies (vdW, hydrogen bonds, and electrostatic) along with the prediction of Gibbs free energy (ΔG), residue propensity and hotspot prediction. Protein-protein interfaces of common origin, function, and composition possess a characteristic proportion of interface energies. Therefore, it is of our interest to examine and compare the percentage contributions of these energies towards interface stabilization in large and small heteromeric interfaces. It was noticed that vdW energy, for the most part, were the predominant energy at the interfaces (~79 %), with Hydrogen Bond energy coming in next at about ~14 %, followed by electrostatic, which contributes the least towards the interface formation (~8 %). Both hydrogen bond and electrostatic energy were seen to be more pronounced at the small interfaces of the protein complexes (Figure 2k and 2l).
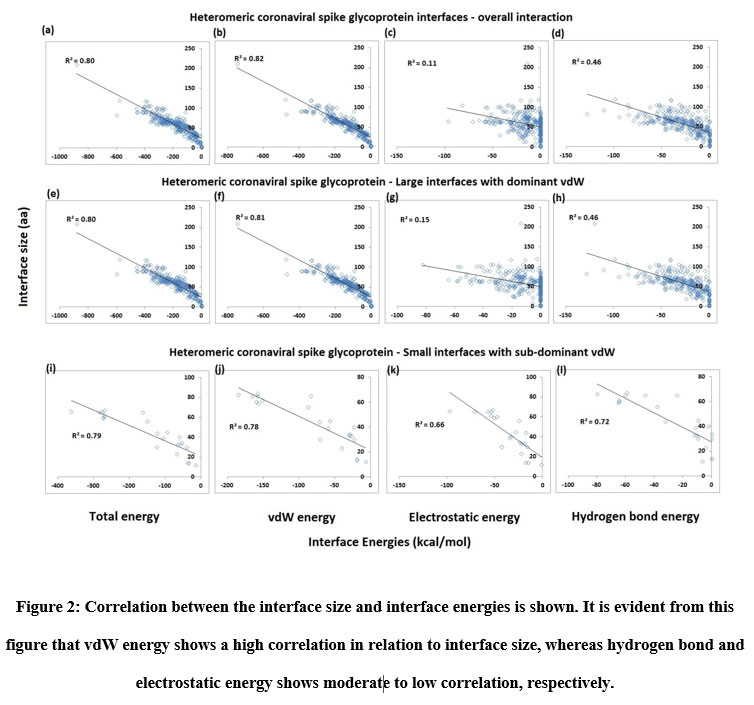 |
Figure 2: Correlation between the interface size and interface energies is shown. It is evident from this figure that vdW energy shows a high correlation in relation to interface size, whereas hydrogen bond and electrostatic energy shows moderate to low correlation, respectively. |
From the figures so far, it is clear that vdW forces reign as the prevailing interaction at the heteromeric interfaces, with limited contributions of hydrogen bond and electrostatic energy. Nevertheless, the presence and contributions of these two interactions cannot be denied and overlooked. So, to ascertain and highlight the importance of hydrogen bond and electrostatic energy interactions, the sub-dominant and dominant vdW categorizations were created for small interfaces. The proportion of vdW energy to total stabilizing energy for sub-dominant vdW interfaces is (≤ 60 %) while that for dominant interfaces is (> 60 %). As per this categorization, about 86% of the total interfaces fall within the dominant vdW category and about 14% of the total interfaces belong to the sub-dominant category. Thus, the partitioning of interfaces as small interfaces with vdW dominant and subdominant aids in discerning those interfaces that exhibit relatively high hydrogen bond and electrostatic energy levels. From Figure 2, it can be noticed that the contribution of hydrogen bond and electrostatic energy to be more significant in the small interfaces of sub-dominant vdW group than vdW dominant group. Furthermore, the small sub-dominant vdW interfaces show an equal degree of hydrogen bond and electrostatic energy contribution, reinforcing the observations in Figure 2k and 2l. The interface area and size of the small interfaces sub-dominant vdW group is smaller than that of the dominant vdW group. The relationship between interface size and energies (vdW, Hydrogen bond, Electrostatics, and total energy) was checked using multiple regression analysis to see the existence of a positive correlation. Figure 2 shows the results of the study carried out on the total interfaces. A robust positive correlation entailing high coefficient of determination values (r2 > 0.8) was seen with vdW and total energy, which is not surprising since vdW forces are non-specific. The atoms need to be nearby for these forces to operate, which occurs when a protein-protein interface manifests. Thus, when the interface size increases, the vdW energy is bound to increase as well. Total energy is just a composite of vdW, hydrogen bond and electrostatic energy, and so, it rises as interface size increases.
 |
Figure 3: The average among the interface area, interface size and interface energy (van der Waals, Electrostatic, Hydrogen bonds) and Gibbs free energy is shown. |
Hydrogen bond and electrostatic energy display a moderate to meager increase with interface size: r2 = 0.46 for the former and r2 = 0.11 for the latter, respectively among the overall interaction including both small and large interfaces. Unlike vdW, hydrogen bond and electrostatic energy that are specific in nature and they require polar or charged residues in the interface. A protein-protein interface rich in serine and threonine or lysine and aspartate would exhibit higher levels of hydrogen bond and electrostatic energy, respectively, than an interface of similar size and area that is rich in non-polar amino acids like valine and alanine. So, these two energies depend just as much, if not more, on the residue composition of the interface as they do on the interface size. With the p-value being (< 0.01) in all the cases, the analyses are statistically significant. For small interfaces, sub-dominant vdW group, the relationship between interface size and vdW and total energy follows a notable positive correlation (Figure 2). Interestingly, the same is observed for hydrogen bond (r2 = 0.72) and electrostatic energy (r2 = 0.66). So, there is a modest increase in these energies with interface size. The analyses are reported to be statistically significant.
Inter-residue analyses were done by computing the Gibbs free energy (ΔG), residue propensity and calculating the interface’s hotspot residues. Residue propensity was computed using an in-house python code, given as supplementary material. In general, we found the interfaces of small and large vdW groups to be equally pronounced with Polar residues. However, we found polar residues to be followed by hydrophobic residues in case of large interfaces and by charged residues in case of small interfaces. Intriguingly, we found small vdW interface group to be devoid of methionine residues (Figure 4). In addition, we also found that most of interfaces to be rich with hotspot residues (Figure 5).
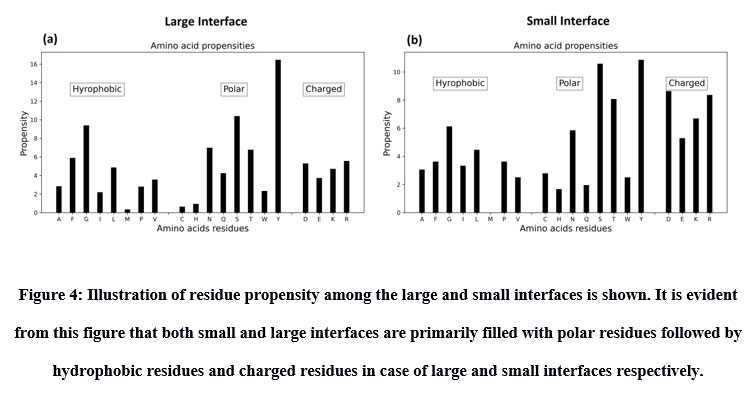 |
Figure 4: Illustration of residue propensity among the large and small interfaces is shown. It is evident from this figure that both small and large interfaces are primarily filled with polar residues followed by hydrophobic residues and charged residues in case of large and small interfaces respectively. |
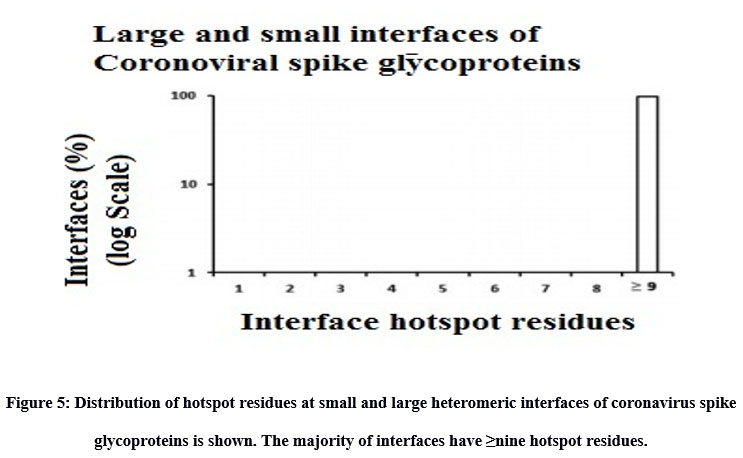 |
Figure 5: Distribution of hotspot residues at small and large heteromeric interfaces of coronavirus spike glycoproteins is shown. The majority of interfaces have ≥nine hotspot residues. |
Discussion
SARS-CoV-2 has incapacitated the health of the human masses and terrified the world’s economy into disorder. We cannot afford to let it or any of its kin hurl in another maelstrom of chaos again. For this reason, there is an urgent need to focus on discovering and bringing new potential remedies to deal with coronavirus. A varied range of solutions has been raised from the persistent efforts of many scientists and researchers across the globe (Odolczyk et al., 2021; Auwaerter & Casadevall, 2020; Ahsan et al., 2020; Yang, 2021). Research into the functioning of SARS-CoV-2 has disclosed enticing new particulars about the linchpins in its viral constitution, which serve as potential drug targets (Sakkiah et al., 2021; Xie et al., 2020; Gordon et al., 2020). Incited by the determined works happening across the globe, this study endeavors to describe the large and small heteromeric interfaces of coronavirus spike glycoproteins, proving to be productive in developing new and novel healing therapies.
Understanding the fundamental driving force of the coronavirus protein interfaces is a crucial step in developing an effectual and efficient therapeutic remedy. This study analyzed 115 of large and 18 of small heteromeric interfaces to discover why the small interfaces of coronaviral heteromeric interfaces are rich in electrostatics; we examined the large and small vdW interfaces using interface size, interface area, interface energy (vdW, hydrogen bond, electrostatic, and total energy), Gibbs free energy (ΔG), residue propensity, and hotspot prediction. The interface area increased with interface size with a high coefficient of determination for small and complete interfaces. The average percentage energy contribution of interface energy reveals that interfaces, on average, are predominant in vdW forces (~ 78 %) with modest contributions of hydrogen bond (~14 %) and electrostatic energy (~ 8 %). Nilofer et al. (2017) reported similar observations in their study of protein-protein interfaces. However, the contributions of hydrogen bond and electrostatic energy are noticeable at small interfaces.
The interfaces are distributed normally, with an increasing percentage of vdW and hydrogen bond energy. This trend isn’t sustained with percentage electrostatic energy, where the interface distribution pattern is reminiscent of a power-law graph. This matches with the observations seen elsewhere (Negi et al. 2007). To shed light on the contributions of hydrogen bond and electrostatic energy, the small interfaces were partitioned into sub-dominant and dominant vdW categories. Consequently, it was seen that 86 % of the total protein-protein interfaces are vdW dominant having large interface, with limited contributions of hydrogen bond and electrostatic energy, whereas 14 % of the interfaces belong to the sub-dominant vdW having small interfaces. The small interfaces sub-dominant vdW group showed exciting results. This group boasts a considerable degree of hydrogen bond and electrostatic energy, with the latter being about three folds higher than the small interfaces dominant vdW group. This outcome is concordant with the observation that small interfaces are richer than large interfaces in charged and polar residues (Kundrotas and Alexov, 2006). It is shown in previous studies that interface energy increases with interface size and interface area (Nilofer et al. 2017; Sowmya and Ranganathan, 2015). The hydrogen bond and electrostatic energy do not increase with interface size for all the groups above, barring the small interfaces sub-dominant vdW class wherein these energies reasonably increase with interface size, on par with the group’s vdW and total energy. This is evidenced by their substantial coefficient of determination values r2: 0.72 for hydrogen bond energy and r2: 0.66 for electrostatic energy.
The analyses show that vdW forces dominate the heteromeric protein-protein interfaces of spike glycoprotein coronavirus. The contributions of hydrogen bond and electrostatic energy are more conspicuous at small interfaces. But what engenders great interest is that the small heteromeric interfaces of coronaviruses having a limited interface area (< 1000 Å2) with sub-dominant vdW energy are sustained primarily by hydrogen bond and electrostatic energy: the former being the greater of the two. Within the 1000 Å2 interface area boundary, these two energies increase with interface size. The inter-residue analysis of the heteromeric interfaces of spike glycoprotein in terms of Gibbs free energy (ΔG), residue propensity and hotspot prediction revealed that the protein interfaces are highly pronounced with polar amino acid residues followed by charged and hydrophobic amino acid residues. This result shows the characteristics of the interface to be biased with polar amino acid residues. Intriguingly, we found small interfaces to be devoid of methionine residues, which trigger our curiosity to research more on the interface region of the small interfaces. It was also found that the small and large interfaces to be populated with normalized energy residues less than one called hotspot residues.
This study has given many compelling outcomes. These intriguing findings are sure to be of immense help in advancing our comprehension of the workings of the protein-protein interfaces of spike glycoprotein coronavirus and building novel counters against them. This, however, does not mark the end of the study. Examining the relationship between the Interface energies calculated using PPCheck and experimentally determined dissociation constants will increase our understanding about the analyses when such data is available for perusal. In addition, the effect of interface water in the estimation of interface energies is neglected in this study. However, future studies could consider the contributions to explore such effects using molecular dynamics simulation in water.
Conclusion
The study aims to compare and analyze the small and large interfaces of heteromeric interactions in coronaviral spike glycoproteins. The small interfaces of coronaviral heteromers were vdW subdominant, with an equal degree of electrostatic and hydrogen bond energy to large interfaces. Gibbs free energy (ΔG) of small interfaces was -30 kcal/mol, whereas it was -22 kcal/mol for large interfaces. Unlike large interfaces, the residue propensities at the small interfaces were found to have polar residues in abundance followed by charged and hydrophobic residues. Interestingly, we found small interfaces to be devoid of Methionine. We also found the small and large interfaces populated with normalized energy residues less than one (<1) termed as hotspot. Thus, this comparative study will help us to glean a deeper understanding of the heteromeric interface interactions of coronavirus spike glycoproteins, proving to be helpful in developing new and productive therapeutic remedies.
Acknowledgement
Christina Nilofer thanks Vikas Tiwari from NCBS for helping with interface energy calculation and hotspot residue analysis using PPCheck. CN thanks Dr. Arnold Emerson for his contribution in developing python script for residue propensity calculation and for his critical comments and corrections on the manuscript. CN thanks Ramanathan Sowdhamini for her continuous support in this PhD journey. CN also thanks Chakravarthy Muralidharan Bhargavan and Navaneethan Nandha for their contribution and support towards this paper.
Conflict of Interest
The authors declare that there is no financial conflict of interest
Funding Sources
The authors declare that there is no funding support for this publication
References
- Ahsan W, Alhazmi HA, Patel KS, Mangla B, Al Bratty M, Javed S et al. Recent Advancements in the Diagnosis, Prevention, and Prospective Drug Therapy of COVID-19. Frontiers in public health 2020;8:384.
CrossRef - Auwaerter PG, Casadevall A. Is the Coronavirus Treatable? Johns Hopkins Medicine. 2020.
- Bahadur, R. P., Chakrabarti, P., Rodier, F., & Janin, J. (2004). A dissection of specific and non-specific protein–protein interfaces. Journal of Molecular Biology, 336(4), 943–955. doi:10.1016/j.jmb.2003.12.073
CrossRef - Bendell, C. J., Liu, S., Aumentado-Armstrong, T., Istrate, B., Cernek, P. T., Khan, S., … Murgita, R. A. (2014). Transient protein–protein interface prediction: Datasets, features, algorithms, and the RAD-T predictor. BMC Bioinformatics, 15(1), 82. doi:10.1186/1471-2105-15-82
CrossRef - Caffrey, D. R., Somaroo, S., Hughes, J. D., Mintseris, J., & Huang, E. S. (2004). Are protein–protein interfaces more conserved in sequence than the rest of the protein surface? Protein Science, 13(1), 190–202. doi:10.1110/ps.03323604
CrossRef - Chakrabarti, P., & Janin, J. (2002). Dissecting protein–protein recognition sites. Proteins: Structure, Function, and Genetics, 47(3), 334–343. doi:10. 1002/prot.10085
CrossRef - Chang CK, Lin SM, Satange R, Lin SC, Sun SC, Wu HY. Targeting protein-protein interaction interfaces in COVID-19 drug discovery. Computational and structural biotechnology journal 2021;19:2246–2255.
CrossRef - Chothia, C., & Janin, J. (1975). Principles of protein–protein recognition. Nature, 256(5520), 705–708. doi:10.1038/256705a0
CrossRef - Chothia, C., Wodak, S., & Janin, J. (1976). Role of subunit interfaces in the allosteric mechanism of hemoglobin. Proceedings of the National Academy of Sciences of the United States of America, 73(11), 3793–3797. doi:10.1073/pnas.73.11.3793
CrossRef - Correa Marrero, M., Immink, R. G. H., de Ridder, D., & van Dijk, A. D. J. (2019). Improved inference of intermolecular contacts through protein–protein interaction prediction using co-evolutionary analysis. Bioinformatics, 35, 2036–2042. doi:10.1093/bioinformatics/bty924
CrossRef - Daberdaku, S., & Ferrari, C. (2018). Exploring the potential of 3D Zernike descriptors and SVM for protein-protein interface prediction. BMC Bioinformatics, 19(1), 35. doi:10.1186/s12859-018-2043-3
CrossRef - Dai, W., Wu, A., Ma, L., Li, Y. X., Jiang, T., & Li, Y. Y. (2016). A novel index of protein–protein interface propensity improves interface residue recognition. BMC Systems Biology, 10(S4), 112. doi:10.1186/s12918- 016-0351-7
CrossRef - Dauzhenka, T., Kundrotas, P. J., & Vakser, I. A. (2018). Computational feasibility of an exhaustive search of side-chain conformations in protein–protein docking. Journal of Computational Chemistry, 39(24), 2012–2021. doi:10.1002/jcc.25381
CrossRef - Garcia-Garcia, J., Valls-Comamala, V., Guney, E., Andreu, D., Munoz, F. J., ~ Fernandez-Fuentes, N., … Oliva, B. (2017). iFrag: A protein–protein interface prediction server based on sequence fragments. Journal of Molecular Biology, 429(3), 382–389. doi:10.1016/j.jmb.2016.11.034
CrossRef - Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, Braberg H. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science (New York, N.Y.), 2020;370:eabe9403.
- Gromiha, M. M., Yokota, K., & Fukui, K. (2009). Energy based approach for understanding the recognition mechanism in protein–protein complexes. Molecular Biosystems, 5, 1779–1786. doi:10.1039/b904161n
CrossRef - Guharoy, M., & Chakrabarti, P. (2005). Conservation and relative importance of residues across protein-protein interfaces. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15447–15452. doi:10.1073/pnas.0505425102
CrossRef - Guharoy, M., & Chakrabarti, P. (2010). Conserved residue clusters at protein–protein interfaces and their use in binding site identification. BMC Bioinformatics, 11(1), 286. doi:10.1186/1471-2105-11-286
CrossRef - Guo, F., Ding, Y., Li, S. C., Shen, C., & Wang, L. (2016). Protein–protein interface prediction based on hexagon structure similarity. Computational Biology and Chemistry, 63, 83–88. doi:10.1016/j.compbiolchem.2016.02.008
CrossRef - Hardcastle IR, Cancer, Immunology and Inflammation, and Infectious Disease. Comprehensive Medicinal Chemistry III. 2017.
- https://covid19.who.int/ (accessed on 20 July 2021)
- https://www.livemint.com/science/health/from-alpha-beta-gamma-to-delta-what-we-know-of-coronavirus-mutations-so-far-11625207686643.html (accessed on 20 July 2021)
- https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 20 July 2021)
- Hubbard S, Thornton J. NACCESS, computer program. London: Department of Biochemistry Molecular Biology, University College. 1993.
- Humphris, E. L., & Kortemme, T. (2008). Prediction of protein–protein interface sequence diversity using flexible backbone computational protein design. Structure, 16(12), 1777–1788. doi:10.1016/j.str.2008.09.012
CrossRef - Hwang, H., Petrey, D., & Honig, B. (2016). A hybrid method for protein–protein interface prediction. Protein Science, 25(1), 159–165. doi: 10.1002/pro.2744
CrossRef - Janin, J., & Chothia, C. (1990). The structure of protein–protein recognition sites. The Journal of Biological Chemistry, 265(27), 16027–16030. Jones, S. (2012). Computational and structural characterisation of protein associations. Advances in Experimental Medicine and Biology, 747, 42–54. doi:10.1007/978-1-4614-3229-6_3
CrossRef - Jones, S., & Thornton, J. M. (1995). Protein–protein interactions: A review of protein dimer structures. Progress in Biophysics and Molecular Biology, 63(1), 31–65. doi:10.1016/0079-6107(94)00008-W
CrossRef - Jones, S., & Thornton, J. M. (1996). Principles of protein–protein interactions. Proceedings of the National Academy of Sciences of the United States of America, 93(1), 13–20. doi:10.1073/pnas.93.1.13
CrossRef - Jones, S., & Thornton, J. M. (1997a). Analysis of protein–protein interaction sites using surface patches. Journal of Molecular Biology, 272(1), 121–132. doi:10.1006/jmbi.1997.1234
CrossRef - Jones, S., & Thornton, J. M. (1997b). Prediction of protein–protein interaction sites using patch analysis. Journal of Molecular Biology, 272(1), 133–143. doi:10.1006/jmbi.1997.1233
CrossRef - Jordan, R. A., El-Manzalawy, Y., Dobbs, D., & Honavar, V. (2012). Predicting protein–protein interface residues using local surface structural similarity. BMC Bioinformatics, 13(1), 41. doi:10.1186/1471-2105- 13-41
CrossRef - Krissinel E and Henrick K. (2007), ‘Inference of macromolecular assemblies from crystalline state.’. J. Mol. Biol. 372, 774-797. https://www.ebi.ac.uk/msd-srv/prot_int/cgi-bin/piserver
CrossRef - Korn, A. P., & Burnett, R. M. (1991). Distribution and complementarity of hydropathy in multisubunit proteins. Proteins, 9(1), 37–55. doi:10. 1002/prot.340090106
CrossRef - Kundrotas PJ, Alexov E. Electrostatic properties of protein-protein complexes. Biophys J. 2006;91:1724-36.
CrossRef - Lee B, Richards FM. The interpretation of protein structures: Estimation of static accessibility. Journal of Molecular Biology 1971;55:379–400.
CrossRef - Li, B., & Kihara, D. (2012). Protein docking prediction using predicted protein–protein interface. BMC Bioinformatics, 13(1), 7.doi:10.1186/1471- 2105-13-7
CrossRef - Li, L., Zhao, B., Cui, Z., Gan, J., Sakharkar, M. K., & Kangueane, P. (2006). Identification of hot spot residues at protein–protein interface. Bioinformation, 1(4), 121–126. doi:10.6026/97320630001121
CrossRef - Li, M., He, Q., Ma, J., He, F., Zhu, Y., Chang, C., … Chen, T. (2019). PPICurator: A tool for extracting comprehensive protein–protein interaction information. Proteomics, 19(4), 1800291.
CrossRef - Lo Conte, L., Chothia, C., & Janin, J. (1999). The atomic structure of protein–protein recognition sites. Journal of Molecular Biology, 285(5), 2177–2198. doi:10.1006/jmbi.1998.2439
CrossRef - Marchetti, F., Capelli, R., Rizzato, F., Laio, A., & Colombo, G. (2019). The subtle trade-off between evolutionary and energetic constraints in protein–protein interactions. The Journal of Physical Chemistry Letters, 10(7), 1489–1497. doi:10.1021/acs.jpclett.9b00191
CrossRef - Miller, S., Lesk, A. M., Janin, J., & Chothia, C. (1987). The accessible surface area and stability of oligomeric proteins. Nature, 328(6133), 834–836. doi:10.1038/328834a0
CrossRef - Moreira, I. S., Koukos, P. I., Melo, R., Almeida, J. G., Preto, A. J., Schaarschmidt, J., … Bonvin, A. M. J. J. (2017). SpotOn: High accuracy identification of protein–protein interface hot-spots. Scientific Reports, 7(1), 8007. doi:10.1038/s41598-017-08321-2
CrossRef - Murakami, Y., & Jones, S. (2006). SHARP2: Protein–protein interaction predictions using patch analysis. Bioinformatics, 22(14), 1794–1795. doi: 10.1093/bioinformatics/btl171
CrossRef - Negi SS, Schein HC, Oezguen N, Power TD, Braun W. InterProSurf: a webserver fro predicting interacting sites on protein surfaces. Bioinformatics 2007; 23:15.
CrossRef - Nilofer C, Sukhwal A, Mohanapriya A, Kangueane P. Protein–protein interfaces are vdW dominant with selective H-bonds and (or) electrostatic towards broad functional specificity. Bioinformation 2017;13:164–173.
CrossRef - Nilofer C, Sukhwal A, Mohanapriya A, Sakharkar MK, Kangueane P. Small protein–protein interfaces rich in electrostatic are often linked to regulatory function. J Biomol Struct Dyn 2020;38,3260–3279.
CrossRef - Odolczyk N, Marzec E, Winiewska-Szajewska M, Poznański J, Zielenkiewicz P. Native Structure-Based Peptides as Potential Protein-Protein Interaction Inhibitors of SARS-CoV-2 Spike Protein and Human ACE2 Receptor. Molecules (Basel, Switzerland) 2021;26:2157.
CrossRef - Pal, A., Chakrabarti, P., Bahadur, R., Rodier, F., & Janin, J. (2007). Peptide segments in protein–protein interfaces. Journal of Biosciences, 32(1), 101–111.
CrossRef - Porter, K. A., Desta, I., Kozakov, D., & Vajda, S. (2019). What method to use for protein-protein docking? Current Opinion in Structural Biology, 55, 1–7. doi:10.1016/j.sbi.2018.12.010
CrossRef - Qiao, Y., Xiong, Y., Gao, H., Zhu, X., & Chen, P. (2018). Protein–protein interface hot spots prediction based on a hybrid feature selection strategy. BMC Bioinformatics, 19(1), 14. doi:10.1186/s12859-018-2009-5
CrossRef - Robert, C. H., & Janin, J. (1998). A soft, mean-field potential derived from crystal contacts for predicting protein-protein interactions. Journal of Molecular Biology, 283(5), 1037–1047. doi:10.1006/jmbi.1998.2152
CrossRef - Sakkiah S, Guo W, Pan B, Ji Z, Yavas G, Azevedo M et al. Elucidating Interactions Between SARS-CoV-2 Trimeric Spike Protein and ACE2 Using Homology Modeling and Molecular Dynamics Simulations. Frontiers in chemistry 2021;8:622632.
CrossRef - Sowmya, G., Anita, S., & Kangueane, P. (2011). Insights from the structural analysis of protein heterodimer interfaces. Bioinformation, 6(4), 137–143
CrossRef - Sowmya G, Ranganathan S. Discrete structural features among interface residue-level classes. BMC Bioinformatics 2015;16:S8.
CrossRef - Sowmya, G., Breen, E. J., & Ranganathan, S. (2015). Linking structural features of protein complexes and biological function. Protein Science, 24(9), 1486–1494. doi:10.1002/pro.2736
CrossRef - Sukhwal A, Sowdhamini R. Oligomerisation status and evolutionary conservation of interfaces of protein structural domain superfamilies. Molecular Biosystems 2013;9:1652–1661.
CrossRef - Sukhwal A, Sowdhamini R. PPCheck: A webserver for the quantitative analysis of protein–protein interfaces and prediction of residue hotspot. Bioinformatics and Biology Insights 2015;9:141–151.
CrossRef - Taechalertpaisarn, J., Lyu, R.-L., Arancillo, M., Lin, C.-M., Perez, L. M., Ioerger, T. R., & Burgess, K. (2019). Correlations between secondary structure- and protein–protein interface-mimicry: The interface mimicry hypothesis. Organic & Biomolecular Chemistry, 17(12), 3267–3274. doi:10.1039/C9OB00204A
CrossRef - Tsai, C. J., Lin, S. L., Wolfson, H. J., & Nussinov, R. (2008). Studies of protein–protein interfaces: A statistical analysis of the hydrophobic effect. Protein Science, 6(1), 53–64. doi:10.1002/pro.5560060106
CrossRef - Wang, W., Yang, Y., Yin, J., & Gong, X. (2017). Different protein–protein interface patterns predicted by different machine learning methods. Scientific Reports, 7(1), 16023. doi:10.1038/s41598-017-16397-z
CrossRef - Xie Y, Karki CB, Du D, Li H, Wang J, Sobitan A et al. Spike Proteins of SARS-CoV and SARS-CoV-2 Utilize Different Mechanisms to Bind With Human ACE2. Frontiers in molecular biosciences 2020;7:591873.
CrossRef - Xu, D., Tsai, C. J., & Nussinov, R. (1997). Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Engineering Design and Selection, 10(9), 999–1012. doi:10.1093/protein/10.9.999
CrossRef - Xue, L. C., Dobbs, D., & Honavar, V. (2011). HomPPI: A class of sequence homology based protein-protein interface prediction methods. BMC Bioinformatics, 12(1), 244. doi:10.1186/1471-2105-12-244
CrossRef - Yang D. Application of Nanotechnology in the COVID-19 Pandemic. International journal of nanomedicine 2021;16:623–649.
CrossRef - Yang, Y., & Gong, X. (2018). A new probability method to understand protein–protein interface formation mechanism at amino acid level. Journal of Theoretical Biology, 436, 18–25. doi:10.1016/j.jtbi.2017. 09.026
CrossRef - Ye T, Zhong Z, García-Sastre A, Schotsaert M, and De Geest BG. Current Status of COVID-19 (Pre) Clinical Vaccine Development. Angewandte Chemie (International ed. in English) 2020;59:18885–18897.
CrossRef - Zhanhua, C., Gan, J. G., Lei, L., Mathura, V. S., Sakharkar, M. K., & Kangueane, P. (2005). Protein subunit interfaces: Heterodimers versus homodimers. Bioinformation, 1(2), 28–39. doi:10.6026/97320630001028.
CrossRef

