
Manuscript accepted on :06-06-2020
Published online on: 13-06-2020
Plagiarism Check: Yes
Reviewed by: Ian Martins
Second Review by: Monique Mancuso
Final Approval by: Dr. Gul Ozcan
Neyder Contreras-Puentes1* and Antistio Alvíz-Amador2
1GINUMED, Faculty of Medicine, Rafael Nuñez Universitary Corporation, Cartagena D.T y C, Colombia
2Pharmacology and Therapeutic Group, University of Cartagena, Cartagena D.T y C., Colombia
Corresponding Author E-mail : neyder.contreras@curnvirtual.edu.co
DOI : https://dx.doi.org/10.13005/bpj/1962
Abstract
Aims: COVID-19 is a global pandemic that has affected around 186 countries in the world, related to clinical signs as fever, cough and pneumonia. The disease is caused by SARS-CoV-2, in the pathophysiology of SARS-CoV-2 it presents the importance of different structural and functional proteins. Some of these mechanisms are based on proteases such as 3CL-PRO and PL-PRO related to the specific cleavage of polypeptides to replication. Materials and Methods: In order to search for alternatives to counteract the virus, computational screening tools have been used, employing molecular docking methodologies through natural ligands, drugs and analogues against SARS-CoV-2 proteases. Subsequently, were tested by ligand-protein interaction employed AutoDock-Vina and PyRx 0.8. Results: From 93 molecules (38 drugs and analogues with antiviral activity and 55 of natural origin with protease inhibitory activity) selected, the ligands with highest affinity indicated to saikosaponin D and SCHEMBL3057328 for 3CL-PRO; Conversely, for PL-PRO were indicated amentoflavone and MK-3207. The presence of potential inhibitors was contrasted with data from previous studies, in which its capacity in vitro and in vivo was determined to inhibit the development of coronavirus. Thus, substantial contributions in silico in the search for promising alternatives of nature and antiviral drugs, which contributes to the validation and establishment of possible candidates for the inhibition of SARS-CoV-2 proteins, favoring the study of new lines of treatments.
Keywords
COVID-19; Drugs; Molecular Docking; Natural Metabolites; SARS-CoV-2
Download this article as:| Copy the following to cite this article: Contreras-Puentes N, Alvíz-Amador A. Virtual Screening of Natural Metabolites and Antiviral Drugs with Potential Inhibitory Activity against 3CL-PRO and PL-PRO . Biomed Pharmacol J 2020;13(2). |
| Copy the following to cite this URL: Contreras-Puentes N, Alvíz-Amador A. Virtual Screening of Natural Metabolites and Antiviral Drugs with Potential Inhibitory Activity against 3CL-PRO and PL-PRO . Biomed Pharmacol J 2020;13(2). Available from: https://bit.ly/2Yx6fQg |
Introduction
SARS-CoV-2, identified in Wuhan (China) is the main etiologic agent causing of COVID-19, characterized by the clinical development of fever, cough, myalgias, dyspnea involving severe acute respiratory syndrome associated pneumonia. The World Health Organization (WHO) has confirmed more than 900,000 cases and a total of deaths that exceed 40,000 globally, making it the most striking pandemic today with an estimated mortality rate of approximately 2.5 %.1
The development of a vaccine and treatment alternatives are insufficient, as they are procedures that require time to achieve efficiency and safety. However, several options can be envisioned to control or prevent emerging COVID-19 infections, including vaccines, monoclonal antibodies, oligonucleotide-based therapies, peptides, interferon and small molecule therapies. Precisely the development of these alternatives could take months or even years.2 Thus, computational chemistry emerges as an element of development of potential drugs of great utility due to its low cost and ease of access to technologies supported by bioinformatics.
Therefore, using molecular screening of small molecules of the receptor ligand coupling type, some pharmacological treatment alternatives corresponding to promising molecules are presented. These molecules that are evaluated in the present study come from natural plant-type products that have been tested as protease inhibitors against viruses such as HIV, influenza, viral hepatitis (HBV and HCV) as well as experience with infections caused by human coronaviruses (Severe Acute Respiratory Syndrome (SARS) and Middle Eastern Respiratory Syndrome (MERS)).2–9 Additionally, some nucleotide-based antiviral agents have also been tested as benchmarks for two viral protease-like proteins, 3CL-PRO and PL-PRO, which are potential action targets for drugs to prevent viral proliferation, and aim of putting before the scientific community these findings given the urgency of the outbreak of COVID-19.
Material and Methods
Preparation of Ligands and Receptors
Previously to reports of activity on SARS-CoV and SARS-CoV-2, a search of potential ligands against viral protease inhibitory activity from natural sources and drugs was performed. 93 molecules were selected (55 of natural source and 38 drugs and analogues) (Supplementary Material- Table 1 and 2). Subsequently, PubChem database was used to download the structural ligands, which were obtained in mol2 format. Then, BIOVIA Discovery Studio version 4.5 and UCSF Chimera version 1.13 software10 was used for structural correction, geometric optimization, hydrogen addition, charge arrangement and ionizable groups. On the other hand, the representative protein structures of the main protease (3CL-PRO) were obtained from Protein Data Bank database, identified with access code: 6LU7 and papain-like protease (PL-PRO), It was obtained by homology using the FASTA sequence condensed in SwissModel database, identified with code: YP_009725299.1.11 Similarly, proteins in PDB format were prepared by adding hydrogen atoms, elimination of solvent (water), and removal ligands using UCSF Chimera version 1.13 software packages and preparing them using the MMFF94 force field.
Molecular Docking
Molecular docking was performed by AutoDock Vina 4.2.1,12 using PyRx 0.8 software graphical interface 13. A virtual screening was implemented to establish the molecules with highest structural affinity against 3CL-PRO and PL-PRO identified in SARS-CoV-2. Therefore, ligands were minimized energetically using the force-field mmff94; using conjugated gradients in 200 steps developed by Open Babel tools14. Proteins and ligands interacted in a grid space of x = 38.47 Å, y = 45.95 Å, z = 40.96 Å for 3CL-PRO and x = 60.69 Å, y = 44.51 Å, z = 32.51 Å for PL-PRO. Then, it was simulated obtaining conformations classified according to affinity energy value and RMSD. The best conformation structures were obtained and converted to PDB format using PyMOL15. The 2017 version of the BIOVIA Discovery Studio visualizer was used in the identification of interaction force and residues.
Pharmacokinetic, Toxicity and Drug-Likeness Prediction
Based at the molecules with best affinity for proteases, a predictive search of the pharmacokinetic, toxicological and drug-likeness properties was performed using the SwissADME and Gusar on-line servers.16
Results and Discussion
The increasing outbreak of SARS-CoV-2 worldwide has been generated an urgent alarm due to the replicative capacity of new agent, which has induced the contagion of more than 1 million individuals, causing mortality about 60.00017. SARS-CoV-2, belonging to β-coronavirus family, characterized by various proteins involved in its infection, such as the Spike protein (S), the membrane protein (M), RNA-directed RNA polymerase (Pol/RdRp), papain-like proteinase (PL-PRO) and main protease (Mpro) or 3C-like protease (3CL-PRO)18–20. Thus, some of the objectives of recent research against the disease have focused at the characterization pharmacological targets of these proteases, which actively participate in the processing of 1ab polyproteins or 1ab replicase by cleavage of the C-terminals in 11 sites, recognizing the sequence [ILMVF]-Q-|-[SGACN], as well as the ability to link to molecules of ADP-ribose-1′-phosphate (ADRP).20 Furthermore, PL-PRO is involved in cleaving replicase polyprotein in N-terminal ends with a deubiquitinating capacity and involving to K48 and K63 residues.21,22 In order to establish the molecular aspects of binding and search for potential candidates against proteases, different metabolites derived from natural products were studied, as well as various drugs and analogues with antiviral activity.
The molecular interactions between 3CL-PRO and PL-PRO with ligands from natural sources, drugs and analogues are revealed in Figures 1 and 2 (A-F), in which are shown the ligands with the highest binding energy, showing interactions common on the active site, interaction residues and type binding force. In Table 1, binding energies of the predominant ligands in natural sources are specified, demonstrating affinity values between -8.6 and -9.2 Kcal/mol for 3CL-PRO; and affinities between -8.5 and -9.2 Kcal/mol for PL-PRO. Likewise, in the Table 1, shows the interaction of higher energy drugs and analogues, showing affinities between -9.4 and -10.2 Kcal/mol for 3CL-PRO and -8.9 and -10 Kcal/mol for PL-PRO. On the other hand, common amino acids located in the binding site are shown as K137, D289 and E290. The highest affinity with 3CL-PRO was demonstrated with saikosaponin D and SCHEMBL3057328, which has shown that these structures interact with K137, V171, A194. PL-PRO showed that the most related molecules were amentoflavone and MK-3207, which interact with P804, R810, V811, A813, T820, P822 and L825. These results are characterized by high hydrophobicity and considerable electron acceptor and donor, with the presence of interactions alkyl, π-alkyl and C-H bonds.
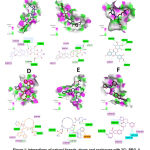 |
Figure 1: Interactions of natural ligands, drugs and analogues with 3CL-PRO. A. 3CL-PRO |
 |
Figure 2: Interactions of natural ligands, drugs and analogues with PL-PRO. A. PL-PRO |
Hence, in silico studies using molecular docking, different promising molecules were shown with representative binding to both proteases such as theaflavin and glycyrrhizin. Myristicrin, saikosaponin A and D were identified for 3CL-PRO; as well as amentoflavone, isoquercitrin and Crisin-7-O-glucuronide for PL-PRO (Table 1). Additionally, drugs and analogues were evaluated in which compounds MK-3207 showed highest affinity for PL-PRO and paritaprevir, SCHEMBL3057328, ledisprevir that evidenced good binding energy in both proteases.
Table 1: Natural ligands, drugs and analogues of higher binding energy as possible 3CL-PRO and PL-PRO inhibitors
| Protein | Ligands | Binding energy (Kcal/mol) | Residues |
| 3CL-PRO | Saikosaponin D | -8.9 | K5, K137, Y239, M276, L286, D289, E290 |
| Saikosaponin A | -8.9 | C128, K137, M276, Y239, M276, L286, D289, E290 | |
| Myricitrin | -8.9 | T26, M49, L141, C145, H163, E166 | |
| Theaflavin | -8.6 | V171, Y237, N238, D289, E290 | |
| Glycyrrhizin | -8.7 | R131, K137, V171, D197, D289, E290 | |
| SCHEMBL3057328 | -10.2 | K137, V171, A194, D197, T199, N238, L286 | |
| Paritaprevir | -10.1 | K137, D197, L286, L287, D289, E290 | |
| MK-3207 | -9.6 | V104, R105, E240, H246, F294 | |
| Ledipasvir | -9.6 | M276, A285, L286, L287, D289, E290 | |
| Velpatasvir | -9.4 | F134, T198, E240, F294 | |
| PL-PRO | Amentoflavone | -9.2 | P804, R810, V811, T820, L825 |
| Glycyrrhizin | -9.6 | H818, N873, Q919, H920, A921 | |
| Theaflavin | -9.1 | P804, R810, A813, F814, D821, P822, L825 | |
| Chrysin-7-O-glucuronide | -8.8 | L907, G928, R911, E912, P993, | |
| Isoquercitrin | -8.5 | P804, R810, A813, P822, L825 | |
| MK-3207 | -10 | P804, R810, A813, F814, T820, D821, P822 | |
| Paritaprevir | -9.8 | P804, R810, V811, A813, F814, P822 | |
| Ledipasvir | -9.2 | T820, H920, L944, M953 | |
| SCHEMBL3057328 | -9.0 | P804, R810, A813, F814, T820, D821, P822, L825 | |
| SCHEMBL1101705 | -8.9 | L803, P804, R810, A813, F814, T820, D821, P822, L825 |
According to the above, recent studies reported that molecules such as saikosaponin A, D and B4 had the ability to bind to the Spike protein of SARS-CoV-2 with binding energies between -11.0 and -13.9 Kcal/mol.23 Likewise, Yan et al, denoted the presence of molecules such as hesperidin, saikosponin, rutin, glycyrrhizin and other compounds against the main protease, with affinities between -8.5 and -8.9 Kcal/mol, similar to reported in this study.24 Similarly, the presence of metabolites with potential inhibitory activity against PL-PRO and 3Cl-PRO have revealed the affinity of cryptotanshinone, quercetin, kaempferol and tanshinone IIa, against both proteases as is reported by Zhang et al 2020.25 Correspondingly, similar reports by Alamri et al, identified that paritaprevir and simeprevir were good candidates as 3CL-PRO inhibitors, with binding energies of -8.8 and 8.78 Kcal/mol.26 Chen et al, performed virtual screenings in which they found that ledipasvir, MK-3207, veltapasvir and other molecules could be potential candidates for SARS-CoV-2 inhibition.27
So, the experimental evidences of these compounds such as saikosponin A and B2 showed in vitro activity against coronaviruses, influencing, in the anchorage, penetration and viral replication against H-Cov-22E9 strains.8 Similarly, glycyrrhizin was evaluated against two strains of SARS-CoV (FFM-1 and FFM-2), which evidenced an potential inhibitor of the replication but low selectivity index.28 Too, Chen et al., showed that theaflavin-derivatives (theaflavin-3,3´-digallate (TF3)) inhibited SARS-CoV 3C-like Protease with values of IC50=7 µM.29 Likewise, the efficacy of ledispavir/sofosbuvir in the inhibition of NS3/4A protease and sustained virological response rate in patients with hepatitis C and HIV have been demonstrated.30 Equally, the efficacy of NS5A inhibitors and polymerase inhibitors by combination of paritaprevir/ritonavir/ombitasvir + dasabuvir or use of ledipasvir/sofosbuvir.31
Conversely, it is distinguished that most of the molecules derived from natural products showed greater affinity for 3CL-PRO, interact with K137, D289 and E290, capable of forming hydrogen bonds with different oxygen of 6-(hydroxylmethyl)oxane-3,4,5-triol and 6-methyloxane, maintaining a polar situation with anchorage site. Furthermore, the drugs and analogues evaluated indicated common residues such as K137 and E290 linked to the oxygens of the structural region of 3,16-diazatricyclo [14.3.0.04,6]nonadec, described in SCHEMBL3057328 and paritaprevir.32 In contrast, the structure of MK-3207 describes the formation of hydrogen bonds with Q110 and E240; as well as, are distinguished interactions between fluorine with R105 and I106. Also, it was identified that natural metabolites linked to 3CL-PRO show a bulky group with little rotation and interconnected hexacyclic, guarantee stability at the binding site, interacting through alkyl or π-alkyl bonds through Y239, M276 and L286.
The analysis of molecular interaction between natural metabolites and PL-PRO, it was shown that amentaflavone, isoquercitrin and theaflavin describe the presence of R810, V811, A813, F814 and L825 that are capable of forming π-alkyl bonds with aromatic rings described in the structures. Moreover, MK-3207 compounds are anchored in binding site by net attractive forces associated to electrophilic region established by fluorine atoms and nucleophilic effect emanating from by A813, T819, D821 and P822.33 Likewise, paritaprevir generates a lipophilic environment with the phenyhridin heterocyclic ring interacting with P804, R810, P822 and L825; and the formation of hydrogen bonds between phenanthridin with T819 and the carbonyl present in 5-methylpyrazine-2-carbonyl with V811.
In the pharmacokinetic and toxicological predictions, it was established that the compound with the best affinity for proteases was MK-3207 (Table 2), where the models showed a good intestinal absorption capacity, do not present permeation to the blood-brain barrier, it’s are characterized by CYP3A4 and 2D6 inhibitors, involved in xenobiotic metabolism that could influence their absorption and ultimately its bioavailability; however, the predictive models used indicated that it has a coefficient of 0.55, which characterizes considerable bioavailability.34 Otherwise, the drug-likeness prediction established that follow the Lipinski, Vogel and Ghose rules with minimal or no violations as established by each of the parameters. Additionally, show toxicity values classify in scale IV, considering slightly toxic and promising molecules as possible inhibitors.
 |
Table 2: Pharmacokinetic, Drug Similarity and Toxicology prediction of potential Inhibitors |
Finally, natural products evaluated in silico such as saikosponin D, amentoflavone and glycyrrhizin have been experimentally tested as antiviral drugs for both current SARS-COV2 and other viruses in general.3,6,7 Saponins such as saikosponin and glycylrrizin seem to be very promising not only because of what has been described in silico but also because of previous reports of their anti-inflammatory and antiviral properties.8, 35
For the other hand, drugs such as ledipasvir, MK-3207 and paratiprevir have been experimentally and even clinically evaluated in the hepatitis C virus.36, 37
Conclusion
From simulations by molecular docking between natural ligands and drugs against 3CL-PRO and PL-PRO, different promising compounds were obtained as potential inhibitors of viral proteases such as saikosponin D, amentoflavone, theaflavin, glycyrrhizin, SCHEMBL3057328, ledipasvir, MK- 3207 and paratiprevir, obtaining the best binding energies and interactions with binding site. The natural product saikosponin and glycylrrizin are they are very promising. Also, pharmacokinetic properties, similarity and toxicity were predicted, in which it was founded that the compound MK-3207 be a promising drug. Finally, it leads to the usefulness of computational tools as an alternative in the selection of possible treatments against COVID-19.
Acknowledgments
Rafael Nuñez University Corporation and University of Cartagena
Funding Source
None
References
- OMS. Coronavirus disease 2019 (COVID-19 ) Situation Report -63. 63, (2020).
- Li, G. & Clercq, E. De. Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nature Reviews Drug Discovery 19, 19–20 (2020).
- Li, S. et al. Identification of natural compounds with antiviral activities against SARS-associated coronavirus. Antiviral Research 67, 18–23 (2005).
- Selvam, P. et al. Anti-influenza virus activities of 4- [( 1 , 2-dihydro-2-oxo- 3H-indol-3-ylidene ) amino ] – N – ( 4 , 6-dimethyl-2-pyrimidin- 2-yl ) benzenesulphonamide and its derivatives. Antiviral Chemistry & Chemotherapy 17, 269–274 (2006).
- Lau, K. et al. Immunomodulatory and anti-SARS activities of Houttuynia cordata. Journal of Ethnopharmacology 118, 79–85 (2008).
- Fu, J., Dai, L., Lin, Z. & Lu, H. Houttuynia cordata Thunb : A Review of Phytochemistry and Pharmacology and Quality Control. Chinese Medicine, 2013, 101–123 (2013).
- Lin, L. T., Hsu, W. C. & Lin, C. C. Antiviral Natural Products and Herbal Medicines. Journal of Traditional and Complementary Medicine 4, 24–35 (2014).
- Cheng, P., Ng, L., Chiang, L. & Lin, C. ANTIVIRAL EFFECTS OF SAIKOSAPONINS ON HUMAN CORONAVIRUS 229E IN VITRO. Clinical and Experimental Pharmacology and Physiology 33, 612–616 (2006).
- Jin, Z., Du, X., Xu, Y., Deng, Y. & Liu, M. Structure-based drug design , virtual screening and high-throughput screening rapidly identify antiviral leads targeting COVID-19. bioRxiv (2020). doi:https://doi.org/10.1101/2020.02.26.964882
- Dassault Systemes BIOVIA. Discovery Studio Visualizer 4.5 (2016). (2016).
- Swiss Institute of Bioinformatics. Severe acute respiratory syndrome coronavirus 2. (2020).
- Trott, O. & Olson, A. J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. Journal of Computational Chemistry 31, 455–461 (2010).
- Sargis, D. & Olson, A. J. Small-molecule library screening by docking with PyRx. in Methods in molecular biology (Clifton, N.J.) 1263, 243–250 (2015).
- Contreras-Puentes, N., Mercado-Camargo, J. & Alvíz-Amador, A. In silico study of ginsenoside analogues as possible BACE1 inhibitors involved in Alzheimer’s disease. F1000Research 8, 1169 (2019).
- DeLano, W. L. PyMOL. 1 (2019).
- Daina, A., Michielin, O. & Zoete, V. SwissADME : a free web tool to evaluate pharmacokinetics , drug- likeness and medicinal chemistry friendliness of small molecules. Scientific Reports 7, 42717 (2017).
- WHO. Coronavirus disease 2019 (COVID-19) . Situation Report – 73. (2020). doi:10.1056/NEJMoa2001316.4.
- Wu, C. et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharmaceutica Sinica B (2020). doi:10.1016/j.apsb.2020.02.008
- Ou, X. et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nature Communications (2019). doi:10.1038/s41467-020-15562-9
- Zhang, L. et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α -ketoamide inhibitors. Science (2020).
- Békés, M. et al. Recognition of Lys48-Linked Di-ubiquitin and Deubiquitinating Activities of the SARS Coronavirus Papain-like Protease. Molecular Cell 62, 572–585 (2016).
- Báez-santos, Y. M., John, S. E. S. & Mesecar, A. D. The SARS-coronavirus papain-like protease : Structure , function and inhibition by designed antiviral compounds. Antiviral Research 115, 21–38 (2015).
- Goswami, T. & Bagchi, B. Molecular Docking study of Receptor Binding Domain of SARS-CoV-2 Spike Glycoprotein with Saikosaponin , a Triterpenoid Natural Product. ChemRxiv. Preprint. (2020). doi:10.26434/chemrxiv.12033774.v1
- Yan, Y.-M. et al. Discovery of Anti-2019-nCoV Agents from Chinese Patent Drugs via Docking Screening. Preprint (2020). doi:10.20944/preprints202002.0254.v1
- Zhang, D. hai, Wu, K. lun, Zhang, X., Deng, S. qiong & Peng, B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. Journal of Integrative Medicine 18, 152–158 (2020).
- Alamri, M. A., Tahir ul Qamar, M. & Alqahtani, S. M. Pharmacoinformatics and Molecular Dynamic Simulation Studies Reveal Potential Inhibitors of SARS-CoV-2 Main Protease 3CLpro. Preprint 1–16 (2020). doi:10.20944/preprints202002.0308.v1
- Chen, Y. W., Yiu, C. B. & Wong, K. Prediction of the SARS-CoV-2 ( 2019-nCoV ) 3C-like protease ( 3CL pro ) structure : virtual screening reveals velpatasvir , ledipasvir , and other drug repurposing candidates [ version 1 ; peer review : 3 approved ]. F1000Research 9, 129 (2020).
- Cinatl, J. et al. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. The Lancet 361, 2045–6. (2020).
- Chen, C. N. et al. Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3,3′- digallate (TF3). Evidence-based Complementary and Alternative Medicine 2, 209–215 (2005).
- Rosenthal, E. et al. Efficacy, safety and patient-reported outcomes of ledipasvir/sofosbuvir in NS3/4A protease inhibitor-experienced individuals with hepatitis C virus genotype 1 and HIV coinfection with and without cirrhosis (ANRS HC31 SOFTRIH study). HIV Medicine 19, 227–237 (2018).
- Janczewska, E. et al. The efficacy of paritaprevir/ritonavir/ ombitasvir+dasabuvir and ledipasvir/ sofosbuvir is comparable in patients who failed interferon-based treatment with first generation protease inhibitors – a multicenter cohort study. BMC Infectious Diseases 2, 1–9 (2018).
- Chandel, V., Raj, S., Rathi, B. & Kumar, D. Approved Antiviral Compounds and Active Phytochemicals through Molecular Docking : Preprint (2020). doi:10.20944/preprints202003.0349.v1
- Cavallo, G. et al. The Halogen Bond. Chemical Reviews 116, 2478–601 (2016).
- Martin, Y. C. A bioavailability score. Journal of Medicinal Chemistry 48, 3164–3170 (2005).
- LuoLiu, P. & Li, J. Pharmacologic perspective: glycyrrhizin may be an efficacious therapeutic agent for COVID-19. International Journal of Antimicrobial Agents 105995 (2020). doi:10.1016/j.ijantimicag.2020.105995
- Bellido-Caparó, Á., Espinoza-Ríos, J. & Tagle Arróspide, M. Tratamiento exitoso con los nuevos antivirales de acción directa en paciente infectado con el virus de la hepatitis C con fibrosis avanzada y dos recaídas previas. Reporte de caso. Revista Medica Herediana 28, 187 (2017).
- AETSA: Agencia de Evaluación de Tecnologías Sanitarias de Andalucía. Ombitasvir / paritaprevir / ritonavir asociado o no , a Dasabuvir en pacientes con Hepatitis C Crónica Eficacia y Seguridad. (2015).

