
Manuscript accepted on :25-03-2020
Published online on: 27-03-2020
Plagiarism Check: Yes
Reviewed by: Shereen fathi
Second Review by: Sujit Nair
Shivani Desai*, Prajakta Pansare, Shivani Sainani, Rohit Doke, Vrushali Bhalchim and Ketki Rode
D. Y. Patil Institute of Pharmaceutical Sciences and Research, Pimpri, Pune 411018
Corresponding Author E-mail : shivani.desai@dypvp.edu.in
DOI : https://dx.doi.org/10.13005/bpj/1897
Abstract
Parkinson is the second most common neurodegenerative disorder that has affected about 6 million people worldwide. The complications of this disorder increase the disability and decrease the QoL of the patient and get worsened as the disease progresses. The therapies available till date have only managed to relieve the suffering along with ample of adverse events they cause. None of the therapy has served to completely cure the disease by targeting its root cause. Being a neurodegenerative disorder, preventing the neurodegeneration and regeneration of neurons might serve to cease the disease progression. Autophagy is a marvelous cleaning mechanism of the body whose impairment is reported to cause accumulation of toxic, misfolded proteins leading to death of neurons. A large number of genes transcription factors and proteins are important players of autophagy which interplay together and cause efficient removal of misfolded, aggregated, toxic proteins to prevent neuronal death. Therefore, these transcription factors may serve as potential targets to trigger autophagy, and thereby prevent neurodegeneration and promote neuroprotection. FoxO6 is known to be one of the important transcription factors in regulating autophagy and hence the aim of the current review is to present a novel strategy for treating PD by triggering autophagy which is in-turn by targeting the concerned genes and transcription factors with a special emphasis on FoxO.
Keywords
FoxO; FoxO6; Neurodegeneration; Neuro-Regeneration; Parkinson
Download this article as:| Copy the following to cite this article: Desai S, Pansare P , Sainani S, Doke R, Bhalchim V, Rode K. Foxo6 – A Novel Target for Parkinson’s Disease. Biomed Pharmacol J 2020;13(1). |
| Copy the following to cite this URL: Desai S, Pansare P , Sainani S, Doke R, Bhalchim V, Rode K. Foxo6 – A Novel Target for Parkinson’s Disease. Biomed Pharmacol J 2020;13(1). Available from: https://bit.ly/3dwJLFv |
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting more than 2% of people over the age of 50. There are more than 6 million cases of PD worldwide [1]. Over the past few years the prevalence of PD has been continuously increasing and is a major contributor to the global disease burden. The exact cause of the disorder is not known but it is assumed to be due to both environmental influences and genetic predisposition [2]. Patients affected with PD suffer from motor and non-motor troubles which critically affect their quality of life (QOL). The motor symptoms of PD are usually encapsulated by ‘TRAP’ meaning, Tremor, Rigidity, Akinesia and Posture instability [3]. Nonmotor symptoms of PD include Dysautonomia, and Neuropsychiatric, Sleep and Sensory complications. As per the Pathological hallmark of PD, loss or degeneration of dopaminergic neurons in the substantia nigra along with deposition of Lewy bodies in the neurons are known to be the pathological reasons [4]. Though Parkinson is known since 18thcentury, curative treatment for the disorder still remains elusive. Although levodopa is considered as the gold standard, its use is associated with motor complications like wearing off, dyskinesia, and on−off phenomenon [5]At the same time, the adverse effects of all the current anti-Parkinson drugs are inevitable [6] and hence there is a strong need of novel therapies for PD. The limited regeneration power of the CNS represents a major challenge for the development of new therapeutic strategies efficacious to promote its functional repair. Among the novel approaches, one of the approaches recently emphasized is Regeneration of Neurons which may be possible by modulation of transcription factors.
Transcription factors are modular proteins with distinct functions contained within defined domains, such as DNA-binding and transactivation of transcription [7]. The forkhead box subfamily “o”, foxo, is one of the larger family of forkhead genes that encode a class of winged helix-turn-helix proteins which act as transcription factors that control homeostasis in response to external influences including variation in growth factor availability and various [8].Foxo proteins were first identified at chromosomal translocation in rhabdomyosarcomas and acute myeloid leukemias [9][10].
The FoxO class of transcription factors consists of four members: FoxO1, 3a, 4 and 6. The alternative names for these genes used in earlier studies were forkhead in rhabdomyosarcoma (FKHR i.e FoxO1), FKHR like 1(FKHRL1 i.eFoxO3a) and Acute-lymphocyticleukaemia-1 fused gene from chromosome X (AFX or Mllt7 i.e.FoxO4)[11]. FoxO1, 3a and 4 are ubiquitously expressed, but between different cell types or organs, the expression level of these FoxOs can differ considerably. For example, FoxO1 is highly expressed in adipose tissue, whereas FoxO4 is highly expressed in muscle and FoxO3a in liver [12]. FoxO6 expression appears restricted to brain [13].
Recently, variations in the foxO1 gene among others, have been implicated in PD. As a part of a widening investigation of the genetic basis of PD, the potential role of foxo in PD has become of interest [14].
Parkinsonism
Parkinson’s Disease is the second most common progressive neurodegenerative disorder resulting from a pathophysiologic loss or degeneration of dopaminergic neurons in the substantia nigra of the midbrain and development of neuronal Lewy Bodies. Parkinson’s disease was first medically described as a neurological syndrome by James Parkinson in 1817[15] written in an essay named ‘An Essay on Shaking Palsy’[16]. Worldwide incidence estimates of Parkinson disease range from 5 to >35 new cases per 100,000 individual’s year analysis rare before 50 years of age, but the incidence increases 5–10‑fold from the sixth to the ninth decade of life. The global prevalence, conservatively estimated at 0.3% overall, likewise increases sharply with age to >3% in those >80 years of age[17]. One main feature of PD is evidence of Lewy bodies which are eosinophilic, cytoplasmic inclusions of fibrillar, misfolded proteins consisting of α-synuclein, parkin, synphilin, neurofilaments and synaptic vesicle proteins. Protein homeostasis is crucial to sustain cellular health and viability in neurons. Therefore, overexpression of α-synuclein inhibits the normal function of inner-mitochondrial membrane-anchored respiratory chain complexes in whole brain of PD patients, but mostly in nigrostriatal neurons. Increased levels of reactive oxygen species (ROS) might be another cause of neuronal death [18] in midbrain and brainstem, olfactory tubercle, cerebral cortex, and elements of the peripheral nervous system. Currently, diagnosis of Parkinson disease is based on clinical features from history and examination, and as the disease progresses it is based on the patient’s response to dopamine agents and development of motor fluctuations.
Therapies: From Beginning Till Date
Medical therapies are the mainstay to fight the symptomatic suffering from PD where both pharmacological and non-pharmacological approaches are currently being applied by the neurologists. Since PD is characterized by multiple disabling symptoms, its best management would be possible by using multi-disciplinary approaches, each one targeting different disability. The first well-established treatment of PD was treating the Parkinsonian tremor by using belladonna alkaloids [19]. Although they cannot replace pharmacological therapies, the non-pharmacological therapies like physical exercise, speech therapy have known to improve QOL of the patients [20]. In-spite of the fact that Levodopa is considered as the gold standard for PD, due to its motor complications, it is prescribed only if the other dopaminergic treatments fail to serve the purpose [21]. However, it is sometimes given as the first-line treatment in older patients as a mono-therapy or in combination with other agents due to its high efficacy and good safety[22]. The dopamine agonists comprising of ergoline and non-ergoline derivatives are mostly advised at the start of the treatment and among two non-ergoline drugs are preferred since they are more efficacious and safer than the ergolines. Mono amino oxidase (MAO-B) inhibitors and catechol ortho methyl transferase (COMT) inhibitors which act by inhibiting dopamine metabolization are other type of agents known for their safety and efficacy and prescribed either singly or as an adjuvant to levodopa or dopamine agonists. But still there are side effects to be considered like tolcapone leads to hepatotoxicity [23]. Apart from dopamine, other neurotransmitters also are potential targets in treating PD. Amantadine, for instance, possibly works by antagonizing N-methyl-D-aspartic acid receptors. However, the role is not clear but despite its unclear mechanism of action, it is recommended for therapy of motor symptoms in young patients and appears to be useful in decreasing levodopa-induced dyskinesias.
The other therapeutic approaches include pump therapy and surgery. Besides its efficacy in motor symptoms, the Levodopa/carbidopa intestinal gel (LCIG) pump is also known to ameliorate non-motor symptoms and QOL of the patient [23]. Apomorphine pump therapy also shows good efficacy for treating motor symptoms in PD. It is characterized by attaining apomorphine plasmatic maximum in less than 10 minutes after subcutaneous application [24]. In recent years, Deep Brain Stimulation (DBS) has emerged as an efficacious therapy for relieving different neurological symptoms using current pulses in different target areas. It was approved in 2002as an adjunctive therapy in reducing motor fluctuation in advanced Parkinson disease. The globus pallidus intern and the subthalamic nucleus are accepted targets for this procedure, with similar improvements in motor function and similar adverse events. The response to deep brain stimulation is equal to the best response of levodopa, but more effective than medical therapy in improving “on” time without troublesome dyskinesias. Deep brain stimulation typically improves levodopa responsive symptoms (e.g., tremor, bradykinesia, rigidity) and on–off fluctuations and dyskinesias whereas impairments in gait, balance and speech are less likely to improve. Patients should be considered for deep brain stimulation only if adequate trials of multiple medications (e.g., levodopa–carbidopa, dopamine agonists, monoamine oxidase B inhibitors and amantadine) have been unsuccessful[25][26].In spite of aforementioned therapies available, none of them is known to cure PD and hence novel approaches targeting the major pathophysiologic mechanisms is a strongly felt need.
Impaired Autophagy in Parkinson
As mentioned earlier, PD being the second most common neurodegenerative disease with a prevalence of 6 million cases which is predicted to increase up to 9 million people worldwide by 2030. Parkinson is said to have a sporadic sub-type since mutation in one of these familial genes (ATP13A2, DJ1, LRRK2, PARK2, PINK1, PLA2G6, SNCA, SYNJ1, UCHL1 and VPS35) is found in only 5–10% of the cases [27]. This serious disorder is characterized by various risk factors like ageing, environmental pollutants, genetic abnormalities, pesticides, metals, rural living, inflammation, head trauma, smoking and alcohol. The pathological hallmarks of PD involve dopaminergic neuronal degeneration in substantia nigra pars compacta, presence of Lewy bodies which are accumulated by α-Synuclein and some cytoplasmic inclusions and it is evidenced that α synuclein pathology and abnormal gene expression are a result of genetic alterations that are reported to be associated.
PARK 1 and 4 genes specifically encode 140 amino acid protein α-synuclein which is predominantly present in presynaptic nerve endings. This protein is involved in various physiological functions such as coupling with lipid rafts which is required for synaptic localization, regulation of presynaptic vesicular pool with emphasis on dopaminergic storage [28], and regulation of lipid metabolism. Due to abnormal mutation in PARK1 and 4 there is overexpression of α synuclein and formation of Lewis bodies which deposit in form of filaments, fibrils and aggregates leading to defective cellular trafficking which needs to be cleaned. Ultimately impaired proteostasis causes altered chaperone mediated autophagy, dopamine mediated toxicity, enhanced sensitivity to oxidative stress and finally neuronal death [29]. PARK2 gene encodes parkin, a RING-finger domain moiety and functioning as an E3 ubiquitin ligase. Parkin interacts with E2, E3, UCHL-1, UCHL-8, SCF like complex, HSP70 and CHIP thereby showing an important role in Ubiquitin proteasome system, mutation of which is thought to be reported into improper ubiquitination of targeted protein for the proteasomal degradation and thus neurotoxic accumulation which will enhance oxidative stress and cause neuronal death [30][31].
PARK5 is confined to encode a highly copious deubiquitinating enzyme UCH-L1 and responsible for hydrolyzing polymeric ubiquitin chains and also plays a key role in dimerization dependent ubiquitination proteasomal ligase action. Hence it possesses both ubiquitin ligase activity as well as hydrolase activity. UCH-L1 acts upon polyubiquitinated proteins to recycle and produce free ubiquitin molecule after their proteasomal degradation. Mutation in hydrolytic activity of UCH-L1 impairs ubiquitin proteasomal system by reducing the availability of free ubiquitin to ubiquitinate the targets and thus potentially accumulate proteins like α-synuclein. Also, even if ubiquitinated, there may be a chance of ubiquitinated protein accumulation since the deubiquitinating action is compromised. And both of these i.e abnormal protein targets and deubiquitinated proteins may induce neuronal cell death [32].PARK6 encodes 81 amino acid protein PINK1 which phosphorylates mitochondrial proteins in relation to cellular stress to prevent mitochondrial dysfunction but inability to phosphorylate proteins leads to mitochondrial dysfunction [33]. Also, PINK1 is an important player of mitophagy and its mutation can hence impair this essential cleaning process confined to removal of damaged mitochondria.DJ-1, can both sense oxidative stress and also has an antioxidant potential. It can be a direct scavenger by deletion of hydrogen peroxide and it offers protection against endoplasmic reticulum stress. DJ-1 possess chaperone and protease activity [34]. Mutation of this DJ-1 is reported in Parkinson which can disturb the normal functions performed by it.
Mitochondrial dysfunction due to complex-I inactivation, increased α-synuclein, nitrosative stress and the latter increases hydrogen peroxide, ROS, Fe3+ production which causes mitochondrial dependent apoptosis [35][36].Increased NOS, ROS cause S-nitrosylation of parkin through its RING domain which inhibit E3 ligase and thus impair parkins ubiquitin ligase activity thereby causing accumulation of proteins and neuronal death[37]. ROS also cause oxidation of dopamine and increased cytosolic DA leading cytotoxicity [38]. This overall molecular alteration pathway leads to degeneration of dopaminergic neuron as well as other catecholamine imbalance which finally propagate to Parkinson’s. Since all the essential players of autophagy like PINK1, Parkin, Ubiquitin Proteasome System are impaired in Parkinson, the neurons are unable to get efficiently cleaned from the aggregated proteins, cytoplasmic inclusions and damaged organelles due to which the condition gets more worsened.
Triggering Autophagy: Novel Strategy in Ameliorating PD
Autophagy is essential for neuronal homeostasis, and its dysfunction has been directly linked to Parkinson’s. Impairment in the induction of autophagy may result in an autophagic flux defect resulting in misfolded protein aggregation. It has been described that alterations in the lysosomal function, such as reduced lysosomal acidification or decreased activity of lysosomal hydrolases, also underlie the failure of autophagic clearance (observed as increased autophagosome accumulation). Defective lysosomal proteolysis may represent a basis for pathogenic protein accumulations and neuronal cell death in PD [39]. Autophagy also has the ability to decrease the accumulation of toxic, aggregate-prone proteins that cause neurodegeneration [40].
Transcription Factor: FOXO6
Transcription factors are modular proteins that regulate gene expression by turning them on and off such that they are rightly expressed in the cells at the right time and in right amount. By regulating the gene expression, they regulate cell growth, cell proliferation and cell death. Amongst the 2600 transcription factors identified in human genome, FOX family is one of the emerging types of transcription factor family that is believed to play an important role in normal physiology and cellular maintenance and hence may serve as an important target in many neurodegenerative diseases, cardiomyopathy, diabetes, immunity and wound healing. The forkhead gene was randomly identified in mutagenesis study of drosophila melanogaster [41] absence of which was found to result in the characteristic “forked head” appearance of gut which looks like spiked head structure [42]. Till now the number of fox genes in varied organisms is not known but it was estimated to be 170 genes from 14 different species. To differentiate them as per the species, mammalian genome contains 44, Drosophila 11, Caenorhabditis elegans 15, and Xenopus 45 genes [43] In humans, about 19 sub-classes of the Fox family have been identified which are ‘Fork head Box’ A to S. The standard nomenclature concerned to fox family involves Fox (forkhead box) as a unified symbol for all members being forkhead helix transcriptional factors. Subclasses are denoted by letter Like FoxA and within each subgroup’s proteins are denoted by a number like FoxA1, where A denotes subgroup and 1 denotes protein. For human all the letters are capitalized like FOXOA1 and in mouse only first letter of gene is capitalized (Foxo1) [44]. The structural feature of fox protein indicates the presence of 110 amino acid and DNA-binding domain (Forkhead domain), corrugated into 3α helics and a flanking domain of two large ‘winged helix’ for interaction with [45]. Despite versatility of this distinctive fox protein their functions have not been fully explored.As there are complex mechanisms involved in the regulation of these proteins, Fox serves contrary role in different parts of body. Members of the Forkhead box-O class of transcription factors are the mammalian orthologue of DAF-16.[46] Caenorhabditis elegans and drosophila melanogaster have one homologue Daf-16 and dFoxO respectively. In Humans, these orthologues are FOXO1, FOXO3, FOXO4 and FOXO6 which are described in Table no. 1
Table 1:
| Protein | Name | Mouse name | Human chromosomal location | Transcriptional activity | Regulation | References |
| FOXO1a | FKHR
(Forkhead in rhabdomyosarcoma) |
Fkhr1, Foxo1a | 13q14.1 | Highly expressed in adipose tissue, spleen, lymph node. | Adipogenesis, glucose homeostasis and insulin sensitivity | (Reyes 201) (Asselin et al., 2004) |
| FOXO3a | FKHRL1, (FKHR-Like protein 1) | Fkhr2, Foxo3a | 5q35.2-3.3
6q21 17p11 |
Expressed in liver and thymus | Apoptosis, autophagy | (Zhou et al., 2007) |
| FOXO4 | AFX (Acute leukemia fusion gene located in chromosome X), AFX1 | afx1, Afxh, foxo4, MIIt7 | Xq13.1 | Nuclei of cells in the mucosa in GIT | Cellular apoptosis | (Salih et al., 2012) |
| FOXO6 | FOXO6 | Foxo6 | 1p34.1 | Restricted to brain | Memory consolidation | (Golson and Kaestner 2016) |
Transcriptional Regulation
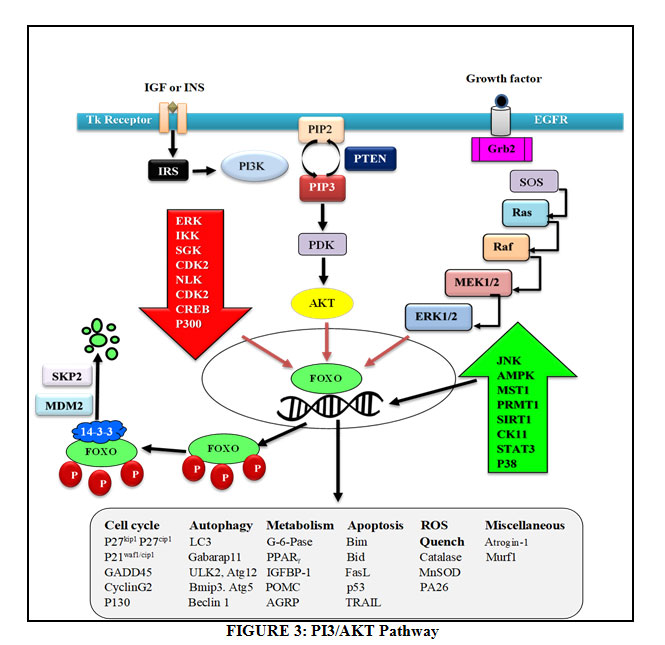
Whenever there are stimuli either for incitement or clampdown the transcription factors are triggered and come into action, they must remain intact to the DNA in nucleus with the transcriptional machinery. In humans there are four distinguishing FoxO members i.e. FOXO1 (FKHR), FOXO3 (FKHRL1), FOXO4 (AFX) and FOXO6. The degree of homology is very high between these four members and possess four discriminate functional elements: Winged helix DNA binding domain (DBD), Nuclear localization Sequence (NLS), Nuclear Export Sequence (NES) and C- terminal transactivation domain. These domains are highly conserved, FoxO3 is significantly larger with 673 amino acids, FoxO1 comprising of 655, FoxO4 is having 505 amino acids and FoxO6 consisting of 559 amino acid sequence. Human FOXO protein are known to be regulated by Phosphatidylinositol 3-kinase/ Protein Kinase B (AKT) [47]. FoxO6, an essential member of the family lacks third conserved PKB Phosphorylation site which is involved in nuclear export of gene and thus residing mostly in nucleus [48]. There are various sites present within these transcriptional factors for Phosphorylation, Acetylation and ubiquitination which regulate post-translational modification as shown in fig.1.
 |
Figure 1: Foxo Genes with Various Domains |
Phosphorylation
PI3K/Akt signal transduction involves phosphorylation of various FoxO proteins. Ak phosphorylates FoxO at three conservative regulatory sites which leads to protein exclusion into the cytoplasm ultimately causing inhibition of target transcriptional gene activity [13]. FoxO6 is exceptional, since only two sites out of 3 are phosphorylated by Akt which is devoid of Nucleocytoplasmic shuttling and its activity is independent of nucleus Exclusion. Various sites of phosphorylation are, for FoxO1, Thr24, Ser256 and Ser319, for FoxO3 Thr32, Ser253 and Ser315, for FoxO4 (Thr28, Ser193 and Ser258) and for FoxO6 Thr26 and Ser 184 (Franke et al., 1997). These various phosphorylation sites lead to sequestration of FoxO to cytoplasm through modulators such as protein kinase B (Akt), Serine/Threonine-protein kinase 1 (SGK1), Casein kinase 1(CK1). Further, post phosphorylation chaperone protein 14-3-3 acts to translocation from nucleus to cytoplasm. [50]. Dual specifity tyrosine-phosphorylation-regulated kinase 1A (DYRK1), Cyclin dependent kinase 1 (CDK 1) results in cytoplasmic accrual. Additionally, inhibitor of nuclear factor kappa-B kinase subunit beta (IKKᵦ) promotes nuclear exclusion and thus stops transcriptional activity, Contrastingly, AMP-Activated protein kinase (AMPK)enhances nuclear translocation by action of Sirt1, a histone deacetylase [51], c-Jun-N-terminal kinases (JNKs) (Pines 1994) and mammalian Ste20-like kinase (Mst1) (Hu MC et al., 2004) promote and augment the transcriptional activity.
Acetylation
Like Phosphorylation, Acetylation also promote suppression as well as augmentation and this activity is regulated by Histone acetyl transferase and Histone deacetylase. FoxO can bind to this enzyme protein which leads to modification. CREB Binding protein (CBP) and p300 are two such types of HAT’s which are requisite for foxO transcriptional activity [53] through acetylation. Several lysine acetylation sites are, for Fox1 K222, K245, K248, K262, K274 and K294 which maintain its sensitivity towards Akt phosphorylation, FoxO3 lysine sites K242, K259, K262, K271, K569 and K290 are acetylated under stress stimuli [54], FoxO4 gets deacetylated by HAT’s ( K186, K189, K408 and K233) and FoxO6 atK173, K176, K190, K202 and K229 [55]. Deacetylation generally favors DNA binding activity and elevates transcriptional activity of target genes, whereas acetylation of foxO itself is known to lessen the transcriptional activity. There are evidences where stress induced acetylation boosts transcriptional activity [56], this is because of complex phenomenon involved in regulation of these protein with respect to acetylation and deacetylation which is unexplored but it is reasserted that phosphorylation and acetylation work conjointly to synchronize FoxO protein.
Methylation
Another post-transcriptional modification involves methylation of arginine and lysine residues at conserved sites of FoxO. Protein arginine methyl transferase PRMT1 methylate foxO at Arg248 and Arg250 which leads to impediment of Akt initiated phosphorylation and thus enhance nuclear localization [57]. Recent affirmation shows that several histone methyltransferase methylate FoxO3 at lysine 270 which leads to decreased DNA binding affinity and termination of transcriptional activity.
Ubiquitination
Whenever there are stimuli for Akt phosphorylation, FoxO is translocated from nucleus to cytoplasm. In fig. 2 it shows that other proteins Phosphorylated FoxO is then ubiquitinated for proteasomal degradation either by Mono-ubiquitination or Poly-ubiquitination. Here skp2 (S-phase kinase -associated protein 2), MDM2 (Mouse double minute 2 homolog) [58], CHIP (carboxy terminus of Hsc70 interacting protein) [59] promotes proteasomal degradation. In case of monoubiquitylation this process is exactly opposite to that described above i.e nuclear localization increases through monoubiquitination thus increasing foxo transcriptional activity. Here binate effects of MDM2 oncogene have also appeared to be a point of interest because monoubiquitnation by MDM2 under oxidative stress pumps the nuclear pool of FoxO and empowers transcriptional activity [60]. Herpes-virus-associated ubiquitin-specific protease, (HAUSP), also known as USP7is a deubiquitinating enzyme exhibiting antagonistic action on FoxO monoubiquitination [61].
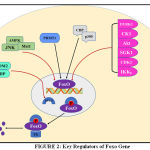 |
Figure 2: Key Regulators of Foxo Gene |
PI3K/AKT Pathway
Foxo regulates PI3K/AKT mTOR pathway which is involved in cell cycle progression which is shown in fig.3 [62] .When cell senses the presence of Insulin , insulin growth factor (IGF), Epidermal Growth Factor (EGF), they bind to the respective receptors and lead to phosphorylation of FOXO.
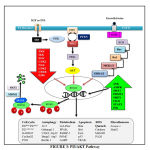 |
Figure 3: PI3/AKT Pathway |
Insulin and IGF activate Insulin Receptor Substrate (IRS) and further PI3K and this activated PI3K converts PIP2 to PIP3 which finally activates Akt. PI3K is antagonized by PTEN. The activated akt phosphorylates foxo at all its 3 different sites of phosphorylation due to which negative charge is induced into its DNA binding domain and due to the negative charged induced, it cannot perform its transcription activities of targeted genes, therefore does not stay at the nuclear region since transcription requires basic charge on the DBD Also, due to the presence of negative charge, it cannot re-enter into the nucleus until it is dephosphorylated again [63]. FoxO1 also get phosphorylated by CDK2 which reduces transcriptional activity of targeted genes and also defeat PTEN-induced activation of FoxO1. In addition to the PI3K/AKT pathway, EGF also phosphorylates FoxO through MAPK signaling pathway which involves activation of ERK 1 and 2 [64].
Talking about its activation and nuclear translocation, it has been discovered that protein phosphatases are responsible for dephosphorylation of FoxO at AKT/SGK sites. But till date, only PP2A is the only one phosphatase that has been characterized to bind FoxO3 [65]. In case of genetic damage, there occurs CHK1/2 mediated translocation of FoxO to the nucleus. In addition to this, oxidative stress causes JNK dependent phosphorylation through MST1 activation, Beta-catenin interaction or direct phosphorylation of FoxO leading to its nuclear translocation. CREB Binding Protein and P300 are histone acetyl transferases which acetylate foxo thereby decreasing its transcriptional activity [66]. On the other hand, its deacetylation by histone deacetylase like SIRT1 boosts its transcriptional activity [67]. Protein Arginine Methyl Transferase (PRMT) 1 and 2 is known to methylate foxo at arginine. But interestingly, it is known that methylation at arginine causes nuclear translocation and hence increases its transcription activity. This is because methylation of arginine compromises akt dependent phosphorylation.
The foxo which is inactivated due to phosphorylation and acetylation is subjected to proteasomal degradation. Therefore, the foxo present in the cytoplasm is targeted by chaperone 14-3-3 for its autophagic removal from the cell. Further, the SKP2 E3 ligase is responsible for the polyubiquitination of foxo after which the ubiquitinated foxo is carried for proteasomal degradation.
Target Genes
As FOXO are the transcriptional regulators, their activation or inhibition leads to expression of certain genes as elaborated in table no. 2
Table 2:
| Sr.no. | Name of gene | Regulator | Activity | Reference |
| 1 | Cellcycle
P27kip1P27cip1
P21waf1/cip1
GADD45
CyclinG2 P130 |
Cyclin E and D Cyclin E and D Miscellaneous
Inhibit E2F4 |
G1 Cell cycle arrest
Cell cycle arrest
Growth arrest and DNA damage repair Arresting cell cycle progression Affect cell cycle regulation |
(Dijkers PF et al., 2000) (Hauck L et al., 2007)
(Tran H et al., 2002)
(Kops GJet al., 2002) |
| 2 | Autophagy
LC3b (Map1lc3b), Gabarapl1, Pi3kIII, Ulk2, Atg12l, Beclin1, Atg4b and Bnip3, Atg5 |
Activate autophagy |
Proteostasis | (Webb AE and Brunet A 2014) |
| 3 | Apoptosis
Bim, Bid,FasL, P53,TRAIL, CHOP |
Inducing expression of death receptor | Apoptosis | (Essafi A et al., 2005),(Urbich C et al., 2005),(Yang JY et al., 2006),(Li Y et al., 2014) |
| 4 | ROS Neutralization
Catalase MnSOD PA26 |
Neutralize hydrogen peroxide to water |
Prevention of cell death |
(Kahl R et al.,2004) |
| 5 | Metabolism
G6Pase,
PPARᵧ, IGFBP-1,
Pomc and Agrp |
Glucose catalytic unit
Insulin response sequence Melanocortin system |
Gluconeogenesis
Adipogenesis Improve insulin sensitivity
Regulation of food intake |
(Gautier Stein A et al., 2012)
(Nunn AV et al., 2007) (Gross DN et al., 2008)
(Sasaki T and Kitamura T 2010) |
| 6 | Angiogenesis
Sprouty2
Angiopoietin (Ang1/2) |
NO synthase
PI3K/Akt |
Negative regulation of Endothelial cell proliferation Modulation of endothelial function |
(Arden KC 2007)
(Shankar S et al., 2008) |
| 7 | Miscellaneous
Atrogin-1
Murf1 |
Myofibril proteins
Proteosomes. |
Skeletal muscle atrophy
Muscle atrophy |
(Sandri M et al., 2004) (Jaitovich A et al., 2015) |
FoxO6
FoxO6 is not only restricted to brain it is also expressed in peripheral tissue including liver, intestine, lung, kidney, muscle and adipose tissue [68]. In brain it is highly expressed in hippocampus, cerebellum, cortex. In adult murine brain’s hippocampus, it is detected in region of dentate gyrus, CA1, CA2 and CA3. In mouse brain it is expressed in a different spatial and temporal manner [69]. As CA1 and CA3 region is involved in learning and memory of adult mice FoxO6 expression thus serves an important role in memory linking [70]. For learning, FoxO6 is activated whenever there is an actual learning task which activates promoter of genes involved in synapse formation [71].
Talking about its similarity to its sibling members, FoxO6 is 34% homologous to Foxo1, 38% homologous to FoxO3, and 36% to FoxO4 for the amino acid length that they are made of. Since FoxO6 is deficient of PKB phosphorylation motif, as discussed earlier, its transcriptional activity regulation is independent of shuttling to cytosol and solely depends on FoxO6-DNA interaction [13]. Two factors i.e. first Ser184 might imply gatekeeper hypothesis and regulates DNA binding while another Thr26 senses growth factor mediated transcriptional regulation. Since it is involved in many important processes of cell biology through its transcriptional activity, FoxO6 is one of the important regulators of oxidative stress and cell proliferation
Foxo in Ameliorating PD
Foxo6 is one of those FOX family members which is discovered to be highly implicated in brain. This is evidenced by the findings of Foxo6 transcript in trigeminal ganglion and in areas around fourth ventricle. To a lesser extent, it was also reported in striatal field of neopallial cortex [69]. It was also eminently expressed in cortical plate of developing hippocampus and striatum [72] . Since it is highly implicated in brain as shown in fig.4 and since there are evidences of Foxo6 being associated with different mechanisms of neuronal regulation and maintenance, there are certain mechanisms through which FOXO is hypothesized to ameliorate PD.
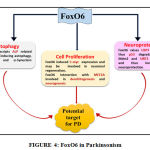 |
Figure 4: FoxO6 in Parkinsonism |
Autophagy
As emphasized continuously, efficient cleaning of every cell by removal of abnormal and damaged moieties is very essential to render it fit and healthy for its survival. It is very well known that neurodegenerative disorders occur due to stress and pollutants characterized by both deficiency in biogenesis of new and healthy proteins and inefficiency in the disposal of expired proteins. The hallmark of PD very well established is the α synuclein pathology combined with damaged organelle accumulation thereby enhancing neuronal stress. Therefore, one of the urgent needs of this abnormality is cleaning of proteins carried out by UPS and ALP and of organelles done by ALP. As mentioned above, FOXO6 is known to transcript Autophagy Related genes like LC3b, Gabarap11, PI3K, ULK2, Atg121, Beclin1, Bnip3, Atg4 and 5 suggesting a role of FoxO in triggering autophagy. Mitochondria is one of the important cell organelles which if damaged, needs to be removed otherwise leads to more production of ROS thereby worsening the condition. FOXO, an emerging transcription factor is known to be involved in inducing mitophagy and general autophagy by transcripting genes known to play these mechanisms. Parkinson is known to be associated with mutation of two important players of mitophagy i.e. PINK1 and parkin thereby indicating impairment of mitophagy in PD. And as emphcized in this review, that FOXO transcripts Atgs, ALP can balance the impaired mitophagy leading to removal of dysfunctional mitochondria along with degradation of α synuclein aggregates and products. Therefore, autophagy can be a potential target of FOXO6 to ameliorate PD.
Neuroprotection
FOXO is also reported to be involved in synapse activity dependent NMDA receptor signaling in neurons. This leads to PI3K/Akt activation which shows neuroprotective action. P53 is one of the important modulators of cell cycle arrest. It is known to activate cyclin dependent kinases by causing their transcription and these kinases like, CIP/WAF1, CDKN1A, Bax are involved in apoptosis [73]. USP7 is another essential molecule that has a role in Ubiquitin Proteosome system. USP7 is known to deubiquitinate Mdm2 rendering it free for action. This Mdm2 is a selective autophagic receptor whose substrate is p53. FoxO6 is known to upregulate USP7 thereby mediating Mdm2 assisted p53 proteosomal degradation [74] . Therefore, there is decrease in apoptosis and hence FoxO6 is thought to play a role in Neuroprotection. Reduced IGF and Insulin is shown to increase life span in C. elegans [75], Drosophila [76] and Rodents through inhibition of apoptosis via SIRT1 action which potentiates cell survival. This occurs through suppression of FoxO and p53 interaction by SIRT [77]. Salih et al have shown FoxO6 to enhance the expression of Trp53 and hence increasing p53 signaling in the hippocampus. This p53 mediated signals are thought to help in synapse formation and memory but the pathway/mechanism involved is burrowed and needs to be digged in to state the explanation with confidence and this will in-turn be possible by extensive study. But this still provides a hope FoxO6 involvement in synapse formation and hence Neuronal Development.
Cell Proliferation
FOXO6 involvement is seen in tumor progression specially in gastric cancer. Therefore, it might play a role in cell proliferation by c-myc expression and may be expected to cause neuronal regeneration.
Also, FOXO6 has an interaction with MEF2A which is known to be associated with neuronal survival and maturation by controlling pre and post synapse formation. It is also known to be involved in dendritogenesis and neurogenesis. Neuronal stem cells are evidenced to be differentiated and matured in the presence of MEF2A (Giannakou ME and Partridge L 2004). Foxo6 may be involved in transactivating several genes that may be required for memory consolidation via MEF2A interaction. But, in contrast, Foxo6 and MEF2A can also show opposing effects (Salih et al., 2012). Foxo is also thought to promote cell survival along with SIRT. Since the above-mentioned evidences and mechanisms indicate the role of FoxO6 in Neuroprotection and Neuronal Development, FOXO6 can be a novel target to ameliorate Parkinson.
Conclusion
Parkinson’s Disorder, being second most common neurodegenerative disorder significantly deteriorates the QoL of the affected patients and causes severe suffering. The treatment available, as known, has numerous side effects. Also, it fails to meet the requirement of eliminating or fighting the actual cause of the neurodegeneration. Therefore, focusing on the increased affectability of population by the disorder it is high time now to bring out an effective treatment that will actually act to cease the neurodegeneration. Since, autophagy is one of the major impairments in Parkinson, triggering autophagy may serve to dispose-off the degraded, misfolded protein aggregates thereby reducing death of neurons. FoxO6 is an astounding molecule that is reported to trigger the transcription of various genes and proteins responsible for autophagy. Hence it might serve as an effective novel target to cure the disorder.
Acknowledgement
The authors are thankful to Dr. D. Y. Patil Institute of Pharmaceutical Sciences and Research, Pimpri, Pune.
References
- Gao, F., Yang, J., Wang, D., Li, C., Fu, Y., Wang, H., … & Zhang, J. (2017). Mitophagy in Parkinson’s disease: pathogenic and therapeutic implications. Frontiers in neurology, 8, 527.
CrossRef - Kieburtz, K., & Wunderle, K. B. (2013). Parkinson’s disease: evidence for environmental risk factors. Movement Disorders, 28(1), 8-13.
CrossRef - Beitz, J. M. (2014). Parkinson Disease : a review. Front Biosci, 6(6574.31).
CrossRef - Macphee, G. J., & Stewart, D. A. (2006). Parkinson’s disease. Reviews in Clinical Gerontology, 16(1), 1-21.
CrossRef - Klivenyi, P., & Vecsei, L. (2010). Novel therapeutic strategies in Parkinson’s disease. European journal of clinical pharmacology, 66(2), 119-125.
CrossRef - Shah, B. (2017). A Review on Pharmacological Treatment of Idiopathic Parkinson’s Disease. Neurol. Disord, 5(3), 1-4.
CrossRef - Burgering, B. T. (2008). A brief introduction to FOXOlogy. Oncogene, 27(16), 2258.
CrossRef - Eijkelenboom, A., & Burgering, B. M. (2013). FOXOs: signalling integrators for homeostasis maintenance. Nature reviews Molecular cell biology, 14(2), 83.
CrossRef - Anderson, M. J., Viars, C. S., Czekay, S., Cavenee, W. K., & Arden, K. C. (1998). Cloning and characterization of three human forkhead genes that comprise an FKHR-like gene subfamily. Genomics, 47(2), 187-199.
CrossRef - Tamai, I., Yabuuchi, H., Nezu, J. I., Sai, Y., Oku, A., Shimane, M., & Tsuji, A. (1997). Cloning and characterization of a novel human pH‐dependent organic cation transporter, OCTN1. FEBS letters, 419(1), 107-111.
CrossRef - Lalmansingh, A. S., Karmakar, S., Jin, Y., & Nagaich, A. K. (2012). Multiple modes of chromatin remodeling by Forkhead box proteins. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms, 1819(7), 707-715.
CrossRef - Furuyama, T., NAKAZAWA, T., NAKANO, I., & Nozomu, M. O. R. I. (2000). Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochemical Journal, 349(2), 629-634.
CrossRef - Jacobs, F. M., Van der Heide, L. P., Wijchers, P. J., Burbach, J. P. H., Hoekman, M. F., & Smidt, M. P. (2003). FoxO6, a novel member of the FoxO class of transcription factors with distinct shuttling dynamics. Journal of Biological Chemistry, 278(38), 35959-35967.
CrossRef - Chavoshi, M. S., & Staveley, B. E. (2016). Inhibition of foxo and minibrain in Dopaminergic Neurons Can Model Aspects of Parkinson Disease in Drosophila melanogaster. Advances in Parkinson’s Disease, 5(01), 1.
CrossRef - Goetz, C. G. (2011). The history of Parkinson’s disease: early clinical descriptions and neurological therapies. Cold Spring Harbor perspectives in medicine, 1(1), a008862.
CrossRef - Jaitovich, A., Angulo, M., Lecuona, E., Dada, L. A., Welch, L. C., Cheng, Y., … & Barreiro, E. (2015). High CO2 levels cause skeletal muscle atrophy via AMP-activated kinase (AMPK), FoxO3a protein, and muscle-specific Ring finger protein 1 (MuRF1). Journal of Biological Chemistry, 290(14), 9183-9194.
CrossRef - Poewe, W., Seppi, K., Tanner, C. M., Halliday, G. M., Brundin, P., Volkmann, J., … & Lang, A. E. (2017). Parkinson disease. Nature reviews Disease primers, 3, 17013.
CrossRef - Chen, X., Guo, C., & Kong, J. (2012). Oxidative stress in neurodegenerative diseases. Neural regeneration research, 7(5), 376.
- Odekerken, V. J., van Laar, T., Staal, M. J., Mosch, A., Hoffmann, C. F., Nijssen, P. C., … & Mink, M. S. (2013). Subthalamic nucleus versus globus pallidus bilateral deep brain stimulation for advanced Parkinson’s disease (NSTAPS study): a randomised controlled trial. The Lancet Neurology, 12(1), 37-44
CrossRef - Ashburn, A., Fazakarley, L., Ballinger, C., Pickering, R., McLellan, L. D., & Fitton, C. (2007). A randomised controlled trial of a home based exercise programme to reduce the risk of falling among people with Parkinson’s disease. Journal of Neurology, Neurosurgery & Psychiatry, 78(7), 678-684.
CrossRef - Pedrosa, D. J., & Timmermann, L. (2013). management of Parkinson’s disease. Neuropsychiatric disease and treatment, 9, 321.
CrossRef - Florian, C., Bahi-Buisson, N., & Bienvenu, T. (2011). FOXG1-related disorders: from clinical description to molecular genetics. Molecular syndromology, 2(3-5), 153-163.
CrossRef - Devos, D., & French DUODOPA Study Group. (2009). Patient profile, indications, efficacy and safety of duodenal levodopa infusion in advanced Parkinson’s disease. Movement Disorders, 24(7), 993-1000.
CrossRef - Stibe, C. M. H., Kempster, P. A., Lees, A. J., & Stern, G. M. (1988). Subcutaneous apomorphine in parkinsonian on-off oscillations. The Lancet, 331(8582), 403-406.
CrossRef - Odekerken, V. J., van Laar, T., Staal, M. J., Mosch, A., Hoffmann, C. F., Nijssen, P. C., … & Mink, M. S. (2013). Subthalamic nucleus versus globus pallidus bilateral deep brain stimulation for advanced Parkinson’s disease (NSTAPS study): a randomised controlled trial. The Lancet Neurology, 12(1), 37-44.
CrossRef - Schuepbach, W. M. M., Rau, J., Knudsen, K., Volkmann, J., Krack, P., Timmermann, L.,& Falk, D. (2013). Neurostimulation for Parkinson’s disease with early motor complications. New England Journal of Medicine, 368(7), 610-622.
CrossRef - Wirdefeldt, K., Adami, H. O., Cole, P., Trichopoulos, D., & Mandel, J. (2011). Epidemiology and etiology of Parkinson’s disease: a review of the evidence. European journal of epidemiology, 26(1), 1.
CrossRef - Bridi, J. C., & Hirth, F. (2018). Mechanisms of α-synuclein induced synaptopathy in Parkinson’s disease. Frontiers in neuroscience, 12, 80.
CrossRef - Moore, D. J., West, A. B., Dawson, V. L., & Dawson, T. M. (2005). Molecular pathophysiology of Parkinson’s disease. Rev. Neurosci., 28, 57-87.
CrossRef - Narendra, D., Tanaka, A., Suen, D. F., & Youle, R. J. (2008). Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. The Journal of cell biology, 183(5), 795-803.
CrossRef - Shimura, H., Hattori, N., Kubo, S. I., Mizuno, Y., Asakawa, S., Minoshima, S., … & Suzuki, T. (2000). Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nature genetics, 25(3), 302.
CrossRef - Maraganore, D. M., Lesnick, T. G., Elbaz, A., Chartier-Harlin, M. C., Gasser, T., Krüger, R., … & Toda, T. (2004). Erratum: UCHL1 is a Parkinson’s disease susceptibility gene (Annals of Neurology (April 2004) 55 (512-521)). Annals of Neurology, 55(6), 899.
CrossRef - Park, J., Lee, S. B., Lee, S., Kim, Y., Song, S., Kim, S., … & Chung, J. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature, 441(7097), 1157.
CrossRef - Zhang, L. I., Shimoji, M., Thomas, B., Moore, D. J., Yu, S. W., Marupudi, N. I., … & Dawson, V. L. (2005). Mitochondrial localization of the Parkinson’s disease related protein DJ-1: implications for pathogenesis. Human molecular genetics, 14(14), 2063-2073.
CrossRef - Kannan, K., & Jain, S. K. (2000). Oxidative stress and apoptosis. Pathophysiology, 7(3), 153-163.
CrossRef - Jenner, P. (2003). Oxidative stress in Parkinson’s disease. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society, 53(S3), S26-S38.
CrossRef - Yao, D., Gu, Z., Nakamura, T., Shi, Z. Q., Ma, Y., Gaston, B., … & Uehara, T. (2004). Nitrosative stress linked to sporadic Parkinson’s disease: S-nitrosylation of parkin regulates its E3 ubiquitin ligase activity. Proceedings of the National Academy of Sciences, 101(29), 10810-10814.
CrossRef - Jana, S., Sinha, M., Chanda, D., Roy, T., Banerjee, K., Munshi, S., … & Chakrabarti, S. (2011). Mitochondrial dysfunction mediated by quinone oxidation products of dopamine: Implications in dopamine cytotoxicity and pathogenesis of Parkinson’s disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease, 1812(6), 663-673.
CrossRef - Lee, J. H., Yu, W. H., Kumar, A., Lee, S., Mohan, P. S., Peterhoff, C. M., … & Uchiyama, Y. (2010). Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell, 141(7), 1146-1158.
CrossRef - Frake, R. A., Ricketts, T., Menzies, F. M., & Rubinsztein, D. C. (2015). Autophagy and neurodegeneration. The Journal of clinical investigation, 125(1), 65-74.
CrossRef - Weigel, D., Jürgens, G., Küttner, F., Seifert, E., & Jäckle, H. (1989). The homeotic gene fork head encodes a nuclear protein and is expressed in the terminal regions of the Drosophila embryo. Cell, 57(4), 645-658.
CrossRef - Pohl, B. S., & Knöchel, W. (2005). Of Fox and Frogs: Fox (fork head/winged helix) transcription factors in Xenopus development. Gene, 344, 21-32.
CrossRef - Wang, D. Y. C., Kumar, S., & Hedges, S. B. (1999). Divergence time estimates for the early history of animal phyla and the origin of plants, animals and fungi. Proceedings of the Royal Society of London. Series B: Biological sciences, 266(1415), 163-171.
CrossRef - Kaestner, K. H., Knöchel, W., & Martínez, D. E. (2000). Unified nomenclature for the winged helix/forkhead transcription factors. Genes & development, 14(2), 142-146.
- Brent, M. M., Anand, R., & Marmorstein, R. (2008). Structural basis for DNA recognition by FoxO1 and its regulation by posttranslational modification. Structure, 16(9), 1407-1416.
CrossRef - Dowell, P., Otto, T. C., Adi, S., & Lane, M. D. (2003). Convergence of peroxisome proliferator-activated receptor γ and Foxo1 signaling pathways. Journal of Biological Chemistry, 278(46), 45485-45491.
CrossRef - Tzivion, G., Dobson, M., & Ramakrishnan, G. (2011). FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1813(11), 1938-1945.
- Wan, H., Dingle, S., Xu, Y., Besnard, V., Kaestner, K. H., Ang, S. L., … & Whitsett, J. A. (2005). Compensatory roles of Foxa1 and Foxa2 during lung morphogenesis. Journal of Biological Chemistry, 280(14), 13809-13816.
CrossRef - Franke, T. F., Kaplan, D. R., & Cantley, L. C. (1997). PI3K: downstream AKTion blocks apoptosis. Cell, 88(4), 435-437.
CrossRef - Essers, M. A., Weijzen, S., de Vries‐Smits, A. M., Saarloos, I., de Ruiter, N. D., Bos, J. L., & Burgering, B. M. (2004). FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. The EMBO journal, 23(24), 4802-4812.
CrossRef - Lehtinen, M. K., Yuan, Z., Boag, P. R., Yang, Y., Villén, J., Becker, E. B., … & Bonni, A. (2006). A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell, 125(5), 987-1001.
CrossRef - Hu, M. C. T., Lee, D. F., Xia, W., Golfman, L. S., Ou-Yang, F., Yang, J. Y., … & Kobayashi, R. (2004). IκB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell, 117(2), 225-237.
CrossRef - Tikhanovich, I., Cox, J., & Weinman, S. A. (2013). Forkhead box class O transcription factors in liver function and disease. Journal of gastroenterology and hepatology, 28, 125-131.
CrossRef - Kobayashi, Y., Furukawa-Hibi, Y., Chen, C., Horio, Y., Isobe, K., Ikeda, K., & Motoyama, N. (2005). SIRT1 is critical regulator of FOXO-mediated transcription in response to oxidative stress. International journal of molecular medicine, 16(2), 237-243.
CrossRef - van der Horst, A., Tertoolen, L. G., de Vries-Smits, L. M., Frye, R. A., Medema, R. H., & Burgering, B. M. (2004). FOXO4 is acetylated upon peroxide stress and deacetylated by the longevity protein hSir2SIRT1. Journal of Biological Chemistry, 279(28), 28873-28879.
CrossRef - Yamagata, K., Daitoku, H., Takahashi, Y., Namiki, K., Hisatake, K., Kako, K., … & Fukamizu, A. (2008). Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Molecular cell, 32(2), 221-231.
CrossRef - Xie, Q., Hao, Y., Tao, L., Peng, S., Rao, C., Chen, H., … & Yuan, Z. (2012). Lysine methylation of FOXO3 regulates oxidative stress‐induced neuronal cell death. EMBO reports, 13(4), 371-377.
CrossRef - Portbury, A. L., Ronnebaum, S. M., Zungu, M., Patterson, C., & Willis, M. S. (2012). Back to your heart: ubiquitin proteasome system-regulated signal transduction. Journal of molecular and cellular cardiology, 52(3), 526-537.
CrossRef - Brenkman, A. B., de Keizer, P. L., van den Broek, N. J., Jochemsen, A. G., & Burgering, B. M. (2008). Mdm2 induces mono-ubiquitination of FOXO4. PloS one, 3(7), e2819.
CrossRef - Nicholson, B., & Kumar, K. S. (2011). The multifaceted roles of USP7: new therapeutic opportunities. Cell biochemistry and biophysics, 60(1-2), 61.
CrossRef - Pópulo, H., Lopes, J. M., & Soares, P. (2012). The mTOR signalling pathway in human cancer. International journal of molecular sciences, 13(2), 1886-1918.
CrossRef - Wallin, J. J., Edgar, K. A., Guan, J., Berry, M., Prior, W. W., Lee, L., … & Salphati, L. (2011). GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Molecular cancer therapeutics, 10(12), 2426-2436.
CrossRef - van der Horst, A., & Burgering, B. M. (2007). Stressing the role of FoxO proteins in lifespan and disease. Nature reviews Molecular cell biology, 8(6), 440.
CrossRef - Mendoza, M. C., Er, E. E., & Blenis, J. (2011). The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends in biochemical sciences, 36(6), 320-328.
CrossRef - Singh, A., Ye, M., Bucur, O., Zhu, S., Tanya Santos, M., Rabinovitz, I., … & Khosravi-Far, R. (2010). Protein phosphatase 2A reactivates FOXO3a through a dynamic interplay with 14-3-3 and AKT. Molecular biology of the cell, 21(6), 1140-1152.
CrossRef - Daitoku, H., Sakamaki, J. I., & Fukamizu, A. (2011). Regulation of FoxO transcription factors by acetylation and protein–protein interactions. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1813(11), 1954-1960.
CrossRef - Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., … & Greenberg, M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. cell, 96(6), 857-868.
CrossRef - Kim, D. H., Zhang, T., Lee, S., & Dong, H. H. (2013). FoxO6 in glucose metabolism. Journal of diabetes, 5(3), 233-240.
CrossRef - Hoekman, M. F., Jacobs, F. M., Smidt, M. P., & Burbach, J. P. H. (2006). Spatial and temporal expression of FoxO transcription factors in the developing and adult murine brain. Gene Expression Patterns, 6(2), 134-140.
CrossRef - Nakashiba, T., Young, J. Z., McHugh, T. J., Buhl, D. L., & Tonegawa, S. (2008). Transgenic inhibition of synaptic transmission reveals role of CA3 output in hippocampal learning. Science, 319(5867), 1260-1264.
CrossRef - Salih, D. A., Rashid, A. J., Colas, D., de la Torre-Ubieta, L., Zhu, R. P., Morgan, A. A. & Madison, D. V. (2012). FoxO6 regulates memory consolidation and synaptic function. Genes & development, 26(24), 2780-2801.
CrossRef - Sun, Z., da Fontoura, C. S., Moreno, M., Holton, N. E., Sweat, M., Sweat, Y., … & Nopoulos, P. (2018). FoxO6 regulates Hippo signaling and growth of the craniofacial complex. PLoS genetics, 14(10), e1007675.
CrossRef - Pollutri, D., Gramantieri, L., Bolondi, L., & Fornari, F. (2016). TP53/microRNA interplay in hepatocellular carcinoma. International journal of molecular sciences, 17(12), 2029.
CrossRef - Hu, H. J., Zhang, L. G., Wang, Z. H., & Guo, X. X. (2015). FoxO6 inhibits cell proliferation in lung carcinoma through up-regulation of USP7. Molecular medicine reports, 12(1), 575-580.
CrossRef - Dillin, A., Crawford, D. K., & Kenyon, C. (2002). Timing requirements for insulin/IGF-1 signaling in C. elegans. Science, 298(5594), 830-834.
CrossRef - Clancy, D. J., Gems, D., Harshman, L. G., Oldham, S., Stocker, H., Hafen, E., … & Partridge, L. (2001). Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science, 292(5514), 104-106.
CrossRef - Giannakou, M. E., & Partridge, L. (2004). The interaction between FOXO and SIRT1: tipping the balance towards survival. Trends in cell biology, 14(8), 408-412.
CrossRef


{kind=link}