
Manuscript accepted on :17-Jan-2020
Published online on: 08-02-2020
Plagiarism Check: Yes
Reviewed by: Monica Butnariu
Second Review by: Saiful Irwan Zubairi
Final Approval by: Dr. Kishore Kumar Jella
Kalpana1, R.K Srivastava2, Ravindra Nath3
1Department of Biotechnology, Dr. Ambedkar Institute of Technology for Handicapped, Kanpur, India
2Rural Division Extension, Central Institute for Medicinal and Aromatic Plant, Lucknow, India
3University Institute of Engineering and Technology, CSJM University,Kanpur,India
Corresponding Author E-mail : kalpna@aith.ac.in
DOI : https://dx.doi.org/10.13005/bpj/1869
Abstract
Cryptosporidium, a member of phylum apicomplexa is considered as an opportunistic pathogen for humans as well as other important livestock. It is a causative agent for water-borne infectious diarrheal like disease cryptosporidiosis with a comparatively high mortality rate among children and immunocompromised patients worldwide. The statics shows that cryptosporidiosis is among the top three threats for the survival of infants, especially in developing countries. To date, no fully effective drug therapy is available to treat cryptosporidiosis. Therefore, the discovery and development of an effective anti-cryptosporidial drug with a novel mechanism of action have become an insistent task for controlling cryptosporidiosis. The literature revealed that various heterocyclic aromatic compounds have various invincible biological properties such as anti-fungal, anti-bacterial and anti-parasitic. Among these, Triazole is one of the most promising candidates that grab attention by researchers, chemists, microbiologists, and pharmacologists through various success stories. Triazole nucleus is present in various natural anti-infective and medicinal compounds. In this research, we have collected triazole compounds from various published works and create a database of these novel compounds for further exploration as anti-cryptosporidial compounds. It is hoped that this research provides new insights for rational anti-cryptosporidial chemotherapeutic agents who will be more active and less toxic.
Keywords
Cryptosporidiosis; IMPDH; Molecular Docking; Triazole; Virtual Screening
Download this article as:| Copy the following to cite this article: Kalpana, Srivastava R. K, Nath R. Comparative Genomics Analysis of Cryptosporidium Parvum and Repurposing of Triazole Derivative as Anti-Cryptosporidial Agents. Biomed Pharmacol J 2020;13(1). |
| Copy the following to cite this URL: Kalpana, Srivastava R. K, Nath R. Comparative Genomics Analysis of Cryptosporidium Parvum and Repurposing of Triazole Derivative as Anti-Cryptosporidial Agents. Biomed Pharmacol J 2020;13(1). Available from: https://bit.ly/2S8kGbl |
Introduction
Cryptosporidiosis is an infectious disease caused by protozoan parasite Cryptosporidium. To date, 27 species and 60 genotypes of the parasite have been identified worldwide. Among them, C. parvum and C. hominis are mainly responsible for human cryptosporidiosis.[1] Human cryptosporidiosis is marked off by greenish watery, profuse fetid diarrhea, dehydration, abdominal pain, fever, and vomiting while failing to gain weight and malnutrition are few symptoms in chronic cases. Immunocompetent host recovers within two weeks in absence of any treatment, but the situation becomes worst in children and host having impaired immune system. In immune-compromised individuals such as those affected by Human Immunodeficiency Virus (HIV) experience unmanageable lethal diarrhea [2]. This acute diarrheal disease remains the largest threat to the health of young children and agriculturally important livestock worldwide [3].The field reports of GEMS (The Global Enteric Multicenter Study ) procured by three-year case-control study over 22000 children of aged five years at seven divergent sites covering Africa and Asia continent manifested parasite second diarrhoea causing agent after rotavirus [4].
Cryptosporidium infection accounts for 20 % and 9 % diarrheal cases in the young population of developing nations and developed nations respectively. In parallel, the findings of Global Network for the study of Malnutrition and Enteric Diseases ( MAL-ED) obtained through a five-year-long birth cohort study in 2145 children of age group 0-24 months at eight public sites in South America, Asia and Africa proclaimed Cryptosporidium species among top five agents that are liable for diarrhoeal mortality in first year of life [4]. In India, the highest occurrence of cryptosporidiosis is noted in southern and northern regions in infants and young children [5].
The cryptosporidium life cycle consisted of both sexual and asexual stages that complete in a single host. Infected hosts shed oocysts in defecation that are infective as well as environmentally resistant phase [6]. Oocysts outlive in extreme environmental conditions and remain unpretentious to chemicals, environmental stresses and even most ultra-water purification drinking and sewage water treatment methods. Pathogenesis begins when oocysts are ingested by the host. Four spindle-like Sporozoites released into the gastrointestinal tract followed by differentiation to trophozoites inside parasitophorous vacuoles that are extracytoplasmic to intestinal epithelial cells. Trophozoites initiate an asexual cycle and undergo two consecutive merogony producing type I and type II meronts. Subsequently, type I merozoites and type II merozoites differentiate from type I and type II meronts respectively. Type I merozoites infect neighbor cells whereas type II merozoites develop into microgamonts (male gametes) and macrogamonts (female gametes) marks the initiation of the sexual phase of reproduction. Than fertilization in between microgametes and macrogametes results in oocysts development. As a result of sporogony, thick-walled oocysts which released in feces and thin-walled oocysts that remain in the host[7].
Now it is established that for such widespread and prevalence of cryptosporidiosis there are mainly two reasons the first one is that infected person sheds large number oocysts that are immediate infective stages to a healthy person. Transmission takes place through a fecal-oral route either directly or indirectly. Another reason is the lack of a fully effective chemotherapeutic agent to treat cryptosporidiosis in all patients [8].
The identification of drug-target enzymes that differ in human counterparts led to the foundation of comparative genomics. Previous studies revealed that Cryptosporidium has many unique metabolic pathways in comparison to other apicomplexan species [9].
However, there is no potential drug/antibiotic against this protozoan disease. The currently available drugs such as decuquinate, sulphaquinoxaline, halofuginone, paromomycin have shown less efficiency. FDA approved nitazoxanide has shown potential effect however in immunodeficiency case it is not effective [10]. Controlling this disease is very difficult because the oocytes of C. parvum are. Thus, it is important to identify the possible drugs to control this life-threatening disease.
Materials and Methods
Retrieval of Proteome and Identification of Non-Homologous Crucial Proteins
Complete genome of C. parvum was retrieved by using the National Center for Biotechnology Information (NCBI) database (http://www.ncbi.nlm.nih.gov/genomes/apicomplexa/) in fasta format[11]. All the sequences manually inspected and short sequences (less than 100 amino acids) were stricken out because short proteins have the least chance to be as essential genes. The CryptoDB (http://CryptpoDB.org) database was chosen to access essential genes of the parasite [12].
Selection of Non-Homologous Proteins
The CD-HIT is a suite of a program that is very useful in comparing and clustering of proteins and nucleotides. The whole proteome of C. parvum was submitted to the CD-HIT server (weizhong-lab.ucsd.edu/cdhit-web-server/cgi-bin/index.cgi?cmd=cd-hit) for filtration of analogous, paralogous and homologous proteins[13]. All the default parameter was chosen except sequence identity cut-off reset to 0.7. The non -homologous proteins of the parasite were obtained through BLAST P analysis as opposed to the human genome with expectation value cut-off reset at 10-4 and other parameters were chosen as default[11]. The non-homologous proteins of the parasite were submitted to the CryptoDB database (http://CryptoDB.org) for the extraction of essential parasite genes. At this step expectation value (E-value) cut-off and bit score cut-off were chosen 10-10 and 100 respectively and the rest of the parameters were chosen as a default[14].
Metabolic Pathway Analysis
KASS-KEGG Automatic Annotation server (http://www.genome.jp/kegg/kass/) was selected for metabolic pathway analysis. KASS program is helpful in the functional annotation of the genome. This analysis results in an assignment of functional role to each gene. This is also helpful in the retrieval of unique proteins for the drug targets[15].
Target Protein Model Generation
Template selection is a process of identifying a suitable protein that shares nearly the same structure of the query protein which doesn’t possess the 3D structure. Template selection is very important in comparative protein modeling. Templates can be chosen by various tools such as BLAST, FASTA, Swiss-model, etc [16]. In the case of BLAST and FASTA, the sequence of the protein in FASTA format can be uploaded and the templates can be manually selected by considering the score value and the E value [17]. In the case of the Swiss-Model server, it automatically chooses the template and models the protein structure.in this study, we choose the BLAST tool for generating a respective template. In this tool, a high level of sequence identity should guarantee a more accurate alignment between the target sequence and template structure [18]. The sequence of CpIMPDH was retrieved from Uniprot and submitted for BLAST against PDB protein for obtaining perspective 3D structure [19]. Hits which fulfilled criteria of query coverage > 95 %, sequence identity >80 % and PDB structure resolution < 3.0 Å were selected as a template for homology modeling of IMPDH
Inosine 5′-monophosphate dehydrogenase (IMPDH) is the most important molecular agent when looking for target-specific drug designing because this protozoan parasite cannot reclaim guanosine and therefore relies on IMPDH for guanine nucleotides synthesis to survive. Interestingly, C. parvum seems to have acquired the IMPDH gene from proteobacteria through lateral gene transfer and IMPDH of C. parvum is functionally and structurally distinct from eukaryotic IMPDH enzymes [20]. Therefore, mutational or inhibitory action against the IMPDH gene is considered to be effective and target specific. In silico identification and optimization of therapeutic inhibitors are cost and time effective. Here most effective additional inhibitors derivatives have been identified and optimized to inactivate the functional domain of IMPDH for treating cryptosporidiosis. Previously we also reported a new chemical scaffold using a structure-based pharmacophore approach against IMPDH of C. parvum.[21]
Active Site Prediction of Target Protein
CASTp 3.0, Computed Atlas of Surface Topography of Proteins (http://www.sts.bioe.uic.edu/castp) is a free online tool which scans structural geometry of macromolecule and shows cavities on physio-chemical properties of residues of surrounding residues[22]. Modeled structure of IMPDH protein submitted to this tool for the spotting of binding pockets. The most prominent binding sites would be selected for further in-silico analysis.
Ligand Data Set Preparation for Virtual Screening
Virtual screening is the foundation pillar for computer-aided drug design paradigm. In this computer-assisted process, virtual evaluation of large chemical libraries is accomplished by docking molecules into target protein and also prioritization of chemical compounds on the basis of binding affinities[23].
In this study, we have chosen the PubChem (https://pubchem.ncbi.nlm.nih.gov) database for finding new molecular scaffolds that selectively interact with the target protein. NCBI operated PubChem database store biochemical information of small molecules under Pccompounds, Pcbioassay, and Pcsubstances subsets[24].
Triazole is five-membered heterocyclic ring compounds with molecular formula C2H3N. The relative positioning of the nitrogen atom in the ring, triazole exists in two isomeric forms and each isomer presents two tautomers. Triazole derivatives interact with biomolecule by forming various noncovalent interactions and thus exploited as medicinal drugs [25]. These compounds were considered for the study.
To date, Pccompounds contain 461,626 triazole compounds, 261, 784 triazole substances and 1507 triazole bioassays. Among 1507 bioassay compounds, a dataset of 30 triazole compounds is prepared by filtering undesirable small fragments, covalent salts, and conformers, etc.
The 2D structures of chemical compounds were drawn in Marvin Sketch[26]. The PDB coordinates of 30 triazole derivatives were obtained after converting their respective mol files in Open Babel[27]. Ligand dataset comprises 30 triazole derivatives[25] and detailed information regarding ligands is provided in Table 1.
Table 1: Triazole Derivate ligands (from PubChem database) for Molecular Docking
| Sl.NO | Compound Name | Compound ID |
| 1 | N-(4-chlorophenyl)-2-(1-naphthalenyloxy)propanamide | S8000001 |
| 2 | N-(4-chlorophenyl)-α-(1-naphthalenyloxy) benzene acetamide | S8000002 |
| 3 | N-(4-chlorophenyl)-3-methyl-2-(1-naphthalenyloxy)butanamide | S8000003 |
| 4 | 2-bromo-N-(4-chlorophenyl)propanamide | S8000004 |
| 5 | N-(4-chlorophenyl)-2-[[4-quinolinyl]oxy]propanamide | S8000005 |
| 6 | 1-[(1-methyl-2-propyn-1-yl)oxy]naphthalene | S8000006 |
| 7 | 1-(1-methylethyl)-2-propyn-1-yl]oxy]naphthalene | S8000007 |
| 8 | 1-(4-chlorophenyl)-4-(2-methyl-1-(1-naphthalenyloxy)propyl)-1H-1,2,3-triazole | S8000008 |
| 9 | 1-(4-chlorophenyl)-4-(1-(naphthalene-1-yloxy)ethyl)-1H-1,2,3-triazole | S8000009 |
| 10 | 1-(2,6-dichlorophenyl)-4-(1-(naphthalen-1-yloxy)ethyl)-1H-1,2,3-triazole | S8000010 |
| 11 | 4-(1-(4-chloronaphthalen-1-yloxy)ethyl)-1-(4-chlorophenyl)-1H-1,2,3-triazole | S8000011 |
| 12 | 4-(1-(1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinoline | S8000012 |
| 13 | 4-((1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)methoxy)quinoline | S8000013 |
| 14 | 4-(1-(1-(3,4-dichlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinolone | S8000014 |
| 15 | 4-(4-(1-(quinolin-4-yloxy)ethyl)-1H-1,2,3-triazol-1-yl)benzonitrile | S8000015 |
| 16 | 2-chloro-4-(4-(1-(quinolin-4-yloxy)ethyl)-1H-1,2,3-triazol-1-yl)benzonitrile | S8000016 |
| 17 | (R)-4-(1-(1-(3,4-dichlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinolone | S8000017 |
| 18 | (S)-4-(1-(1-(3,4-dichlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinolone | S8000018 |
| 19 | (R)-2-chloro-4-(4-(1-(quinolin-4-yloxy)ethyl)-1H-1,2,3-triazol-1-yl) benzonitrile | S8000019 |
| 20 | (S)-2-chloro-4-(4-(1-(quinolin-4-yloxy)ethyl)-1H-1,2,3-triazol-1-yl)benzonitrile | S8000020 |
| 21 | (R)-5-(1-(1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinolone | S8000021 |
| 22 | 4-(1-(1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinoline 1-oxide | S8000022 |
| 23 | 4-(1-(1-(3,4-dichlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinoline 1-oxide | S8000023 |
| 24 | 5-(1-(1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinoline 1-oxide | S8000024 |
| 25 | (R)-4-(1-(1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinoline 1-oxide | S8000025 |
| 26 | (R)-5-(1-(1-(4-chlorophenyl)-1H-1,2,3-triazol-4-yl)ethoxy)quinoline 1-oxide | S8000026 |
| 27 | (R)-5-(but-3-yn-2-yloxy)quinoline 1-oxide | S8000027 |
| 28 | ethyl α-bromocyclopropaneacetate | S8000028 |
| 29 | 2-cyclopropyl-2-(1-naphthalenyloxy)acetic acid | S8000029 |
| 30 | N-(4-chlorophenyl)-2-cyclopropyl-2-(1-naphthalenyloxy) acetamide |
S8000030 |
The PDB coordinates of ligand were converted into pdbqt format after the addition of Gasteiger charges, merging non-polar hydrogens, detecting aromatic carbons and setting up a torsion tree.
Molecular Docking Studies
Molecular docking studies provide significant insights into binding interactions of the ligand with the target protein. AutoDock 4.2 tools were employed for this study[28]. This tool performs rigid docking where ligands and targets are allowed to interact in a rigid state without bond angle, bond length as well as torsional angle alteration.
Result
Metabolic pathway analysis and selection of essential protein in C. parvum
C.parvum has eight chromosomes with 10.4 Mb genome ( Mbp) size that codes for total 18538 proteins sequences[29]. Parasite lacks several key enzymes in comparisons with the host for example it scavenges or transport building block biomolecule from the host in spite of de novo biosynthesis. The whole-genome sequence is manually curated for any observable anomaly CD-HIT (http://weizhong-lab.ucsd.edu) suite of the programme is used for redundancy removal and non -paralogs proteins identification. the whole genome in FASTA format is submitted to cd-hit (http://weizhong-lab.ucsd.edu/cdhit_suite/cgi-bin/index.cgi?cmd=cd-hit) non-homologous proteins were again subjected to BLAST P analysis against the Crypto DB database with 10-10 E-value threshold. This resulted in 323 essential proteins that are described as an essential protein for the propagation of parasite inside the host.
Metabolic Pathway Analysis in Cryptosporidium Parvum
Metabolic pathway analysis shows that parasite has intensely diminished metabolic machinery. Further analysis also suggests that parasite has a scarcity of apicoplast and mitochondrial proteins. Carbohydrates, amino acids, and nucleic acid anabolic functions are also reduced. Parasite depends preponderatingly on glycolysis for energy production. Parasite metabolic machinery perfectly matches with its unique life cycle.
Table 2: List of essential protein of Cryptosporidium parvum which could be a promising target for drug development
| Target Enzyme | Function / Pathway in which the target enzyme is involved |
| Mucin- like proteins | Parasite-host cell interaction and immune invasion |
| Glycosylphosphatidyl-inositol anchored proteins | Pathogenesis |
| N-acetylglucosaminyl transferases | Fatty acid biosynthesis |
| P-type ATPase (CpATpase) | Transporting cations |
| Thrombospondin-related adhesive proteins (TRAPs) | Parasite gliding motility and host-parasite interaction |
| Lactate Dehydrogenase (LDH) | Energy production |
| Malate Dehydrogenase (MDH) | Energy production |
| Inosine Monophosphate Dehydrogenase (CpIMPDH) | Purine and pyrimidine biosynthesis |
| Thymidine Kinase (TK) | DNA Biosynthesis |
| Polyamine enzymes | Polyamine Biosynthesis |
| Dihydrofolate reductase-thymidylate synthetase | Folate Biosynthesis Pathway |
| Cryptosporidium Calcium-dependent protein kinase (CpCDPK1) | Host Cell invasion |
| Clan CA cysteine protease | Host cell invasion |
Target Protein Model Generation
In the results of the BLAST search against PDB, only one-reference protein 3FFS has a high level of sequence identity and the identity of the reference protein with the domain is 99%[16].
 |
Figure 1: BLAST result with a similar template having 99% identity with Inosine Monophosphate dehydrogenase. |
After this, we have chosen 3FFS (PDB ID) as a reference structure for modeling the Inosine Monophosphate dehydrogenase domain. Coordinates from the reference protein (3FFS) to the structurally variable regions (SVRs), structurally constant regions (SCRs), C-termini and C-termini were assigned to the target sequence based on the satisfaction of spatial restraints[30].
The sequence of the reference structures was extracted from the respective structure files and aligned with the target sequence using the default parameters in ClustalW.
The Cladogram tree between the Inosine Monophosphate dehydrogenase and template are at a close distance 0.33 indicates both are closely related in origin[31].
 |
Figure 2: Alignment of Inosine Monophosphate dehydrogenase with template 3FFS. |
The 3FFS structure was used as the templates for building the 3D model of the Inosine Monophosphate dehydrogenase using MODELLER9V7[32].
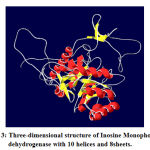 |
Figure 3: Three-dimensional structure of Inosine Monophosphate dehydrogenase with 10 helices and 8sheets. |
The peptide bond of a polypeptide chain N-Cα (phi angle) and Cα-C (psi angle) bond remain free to rotate whereas bond between Cβ-N (omega angle) remain rigid due to π-π interaction. Although the value of phi and psi angle ranges from -180°C to +180°C but because of steric hindrances, only a few limited values are allowed. These dihedral angles describe specific secondary conformation of the protein. Ramachandran Plot helps
in determining secondary structure and assists in structure prediction simulations[33].
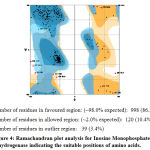 |
Figure 4: Ramachandran plot analysis for Inosine Monophosphate dehydrogenase indicating the suitable positions of amino acids. |
The final structure was further checked by the verify3D graph and the results have been shown in Figure 6. The overall scores indicate an acceptable protein environment.
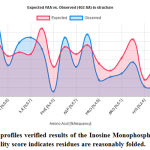 |
Figure 5: The 3D profiles verified results of the Inosine Monophosphate dehydrogenase model; overall quality score indicates residues are reasonably folded. |
After the refinement process, validation of the model was carried out using Ramachandran plot calculations computed with the PROCHECK program[34]. The ψ and ϕ dihedral angle distributions of the Ramachandran plots of non-glycine, non-proline residues are in the permissible range. The RMSD (Root Mean Square deviation) deviation for covalent bonds and covalent angles relative to the standard dictionary of Inosine Monophosphate dehydrogenase was –0.4 and -0.8 Å. Altogether 95% of the residues of Inosine Monophosphate dehydrogenase was in favored and allowed regions. The overall PROCHECK G-factor of Inosine Monophosphate dehydrogenase was – 2.15 and the verify3D environment profile was good.
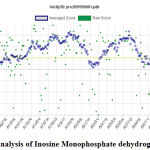 |
Figure 6: Verify 3D analysis of Inosine Monophosphate dehydrogenase. |
The structural superimposition of the 3FFS template and Inosine Monophosphate dehydrogenase is shown in Figure 8. The weighted root mean square deviation of trace between the template and final refined models is 0.41Ao. This final refined model was used for the identification of the active site with the domain Inosine Monophosphate dehydrogenase.
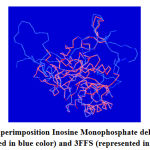 |
Figure 7: Superimposition Inosine Monophosphate dehydrogenase (represented in blue color) and 3FFS (represented in red color). |
Since Inosine Monophosphate dehydrogenase and the 3FFS are well conserved in both sequence and structure; their biological function should be identical. In fact from the structure comparison of template, a final refined model of Inosine Monophosphate dehydrogenase domain using SPDBV program and was shown in Figure 8.
 |
Figure 8: amino acid residues in the active site domain of C.parvum IMPDH shown in red color. |
Active Site Identification
The predicted model was submitted to the CASTp tool for the exploration of major binding pockets. Top three binding pockets in terms of surface area and volume selected. The details of these potential binding sites are given in supplementary table number 1shown in figure 9. We observed that THR 11,PHE12,GLU13, SER22,LEU25,SER 48,ALA49,MET50,ASP163,SER164,ALA165,HIS166, SER169,ASN191,VAL193,LYS210,GLY212,ILE213,VAL214,VAL229,GLN231,ALA234,ASP252,ARG256,TYR257 and ASP260 are binding cavity residues.
 |
Figure 9: Active site identification in Cryptosporidium parvum IMPDH. Molecular Docking |
AutoDock 4.2 a freely accessible computational tool for docking of small molecules to target receptor macromolecule. Bound water molecules and heteroatoms were removed from the target protein. Further, Polar hydrogens and rotatable bonds were selected. This step was followed by the computation of the Gasteiger charge. Target protein preparation was accomplished by choosing amino-acid residues in the active site based on the AutoGrid method. Amino acid residues present in the binding sites include Ser164, Ala165, Ser169, His166, Ser22, Val24, Leu25, Pro26, Asn171, Asn191, and Asp252. Binding site residues were selected and three-dimensional grid boxes were created.
AutoDock exploits the Lamarckian-Genetic algorithm for the accurate placement of ligand into the active site. Docking was executed by considering all stereochemical configurations of chemical compounds. Top 100 poses of ligands were considered for docking in order to enhance the accuracy and efficacy.
Information regarding docking scores is given in Table 2 in ascending order of free binding energy.
Table 3: Molecular Docking result (in term of free binding energy) of Triazole derivative with IMPDH protein of C.parvum
| S. No | Compound ID | Free Binding Energy (Kcal/mol) | S. No | Compound ID | Free Binding Energy (Kcal/mol) |
| 1 | S8000011 | -12.19 | 16 | S8000018 | -10.83 |
| 2 | S8000022 | -12.06 | 17 | S8000019 | -10.54 |
| 3 | S8000012 | -11.7 | 18 | S8000026 | -10.34 |
| 4 | S8000024 | -11.95 | 19 | S8000005 | -10.2 |
| 5 | S8000021 | -11.66 | 20 | S8000008 | -9.9 |
| 6 | S8000020 | -11.6 | 21 | S8000001 | -9.17 |
| 7 | S8000013 | -11.42 | 22 | S8000029 | -7.18 |
| 8 | S8000009 | -11.4 | 23 | S8000030 | -7.08 |
| 9 | S8000025 | -11.29 | 24 | S8000004 | -6.79 |
| 10 | S8000015 | -11.29 | 25 | S8000003 | -6.66 |
| 11 | S8000023 | -11.17 | 26 | S8000002 | -6.24 |
| 12 | S8000016 | -11.12 | 27 | S8000027 | -5.72 |
| 13 | S8000010 | -10.94 | 28 | S8000006 | -5.2 |
| 14 | S8000017 | -10.09 | 29 | S8000007 | -2.4 |
| 15 | S8000014 | -10.83 | 30 | S8000028 | 0.1 |
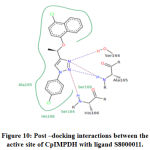 |
Figure 10: Post –docking interactions between the active site of CpIMPDH with ligand S8000011 |
Discussion
IMPDH is an attractive target for the treatment of C. parvum infections. In the present study, we have analyzed the efficacy of 30 triazole derivatives in combating C. parvum infections by executing in-silico methodologies. Molecular docking studies were carried out to identify the efficacy of 30 triazole compounds with CpIMPDH protein. Amino acid residues present in the binding sites include Ser164, Ala165, Ser169, His166, Ser22, Val24, Leu25, Pro26, Asn171, Asn191, and Asp252. The results of the docking study revealed significant interactions of triazole derivatives with IMPDH. Among 30 compounds, 4-(1-(4-chloronaphthalen-1-yloxy) ethyl)-1-(4-chlorophenyl)-1H-1,2,3-triazole (compound ID: S8000011) showed highest free binding energy of -12.19 kcal/mol. This result is coherent with the interactions made by previously reported inhibitors with CpIMPDH. From the docking results, it is evident that chemical compounds possessing both electron releasing and electron-withdrawing groups exhibited good free binding energy values. The presence of electron-withdrawing groups enhances H-bonding potential When the number of electrons withdrawing groups are dominant in the chemical structure, electrons from these groups will be utilized for stabilizing chemical compounds by resonance stabilization interactions. Hence, these electrons can’t be used in making significant interactions with the target protein. This is evident in ethyl α-bromocyclopropaneacetate (Compound ID: S8000011), which exhibited the lowest free binding energy.
Conclusion
The binding efficacies of 30 triazole derivatives, with IMPDH, were analyzed by executing docking studies. Among 30 compounds 4-(1-(4-chloronaphthalen-1-yloxy)ethyl)-1-(4-chlorophenyl)-1H-1,2,3-triazole (Compound ID: S8000011) showed higher free binding energy score with significant interactions with the target protein. Hence, this could be a promising antagonist for IMPDH. But the efficacy of this compound as IMPDH inhibitor has to be proven by conducting in -vitro and in-vivo studies.
References
- L. Xiao, R. Fayer, U. Ryan, and S. J. Upton, “Cryptosporidium taxonomy: recent advances and implications for public health.,” Clin. Microbiol. Rev., 2004.
- L. Xiao, “Molecular epidemiology of cryptosporidiosis: An update,” Exp. Parasitol., 2010.
- S. Bhalchandra, D. Cardenas, and H. D. Ward, “Recent Breakthroughs and Ongoing Limitations in Cryptosporidium Research,” F1000Research, 2018.
- D. A. Shoultz, E. L. de Hostos, and R. K. M. Choy, “Addressing Cryptosporidium Infection among Young Children in Low-Income Settings: The Crucial Role of New and Existing Drugs for Reducing Morbidity and Mortality,” PLoS Negl. Trop. Dis., 2016.
- J. A. Platts-Mills et al., “Pathogen-specific burdens of community diarrhoea in developing countries: A multisite birth cohort study (MAL-ED),” Lancet Glob. Heal., 2015.
- D. P. Clark, “New insights into human Cryptosporidiosis,” Clinical Microbiology Reviews. 1999.
- G. J. Leitch and Q. He, “Cryptosporidiosis-an overview,” Journal of Biomedical Research. 2011.
- N. F. Rossle and B. Latif, “Cryptosporidiosis as threatening health problem: A review,” Asian Pacific Journal of Tropical Biomedicine. 2013.
- H. Sparks, G. Nair, A. Castellanos-Gonzalez, and A. C. White, “Treatment of Cryptosporidium: What We Know, Gaps, and the Way Forward,” Current Tropical Medicine Reports. 2015.
- M. M. Cabada and A. C. White, “Treatment of cryptosporidiosis: Do we know what we think we know?,” Current Opinion in Infectious Diseases. 2010.
- D. L. Wheeler et al., “Database resources of the National Center for Biotechnology Information,” Nucleic Acids Res., 2008.
- M. Heiges, “CryptoDB: a Cryptosporidium bioinformatics resource update,” Nucleic Acids Res., 2006.
- Y. Huang, B. Niu, Y. Gao, L. Fu, and W. Li, “CD-HIT Suite: A web server for clustering and comparing biological sequences,” Bioinformatics, 2010.
- A. Morris, J. Pachebat, G. Robinson, R. Chalmers, and M. Swain, “Identifying and resolving genome misassembly issues important for biomarker discovery in the protozoan parasite, cryptosporidium,” in BIOINFORMATICS 2019 – 10th International Conference on Bioinformatics Models, Methods and Algorithms, Proceedings; Part of 12th International Joint Conference on Biomedical Engineering Systems and Technologies, BIOSTEC 2019, 2019.
- H. Ogata, S. Goto, K. Sato, W. Fujibuchi, H. Bono, and M. Kanehisa, “KEGG: Kyoto encyclopedia of genes and genomes,” Nucleic Acids Research. 1999.
- S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman, “Basic local alignment search tool,” J. Mol. Biol., 1990.
- W. R. Pearson, “Finding protein and nucleotide similarities with FASTA,” Curr. Protoc. Bioinforma., 2016.
- N. Guex and M. C. Peitsch, “SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling,” Electrophoresis, 1997.
- A. Bateman et al., “UniProt: The universal protein knowledgebase,” Nucleic Acids Res., 2017.
- L. Hedstrom, G. Liechti, J. B. Goldberg, and D. R. Gollapalli, “The Antibiotic Potential of Prokaryotic IMP Dehydrogenase Inhibitors,” Curr. Med. Chem., 2011.
- R. K. Srivastava and R. Nath, “Structure based drug designing against Inosine Monophosphate Dehydrogenase Receptor of Cryptosporidium parvum,” in 2018 International Conference on Bioinformatics and Systems Biology (BSB), 2018, pp. 128–130.
- W. Tian, C. Chen, X. Lei, J. Zhao, and J. Liang, “CASTp 3.0: computed atlas of surface topography of proteins.,” Nucleic Acids Res., 2018.
- L. G. Ferreira, R. N. Dos Santos, G. Oliva, and A. D. Andricopulo, “Molecular docking and structure-based drug design strategies,” Molecules. 2015.
- S. Kim et al., “PubChem substance and compound databases,” Nucleic Acids Res., 2016.
- S. K. Maurya et al., “Triazole inhibitors of Cryptosporidium parvum inosine 5′- monophosphate dehydrogenase,” J. Med. Chem., 2009.
- ChemAxon, “Marvin Sketch,” https://www.chemaxon.com/products/marvin/, 2013. .
- P. W. Rose et al., “The RCSB protein data bank: Integrative view of protein, gene and 3D structural information,” Nucleic Acids Res., 2017.
- W. Forli, S. Halliday, R. Belew, and A. Olson, “AutoDock Version 4.2,” Citeseer, 2012.
- M. S. Abrahamsen et al., “Complete Genome Sequence of the Apicomplexan, Cryptosporidium parvum,” Science (80-. )., 2004.
- N. N. Umejiego, C. Li, T. Riera, L. Hedstrom, and B. Striepen, “Cryptosporidium parvum IMP dehydrogenase: Identification of functional, structural, and dynamic properties that can be exploited for drug design,” J. Biol. Chem., 2004.
- J. D. Thompson, D. G. Higgins, and T. J. Gibson, “CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice,” Nucleic Acids Res., 1994.
- J. A. R. Dalton and R. M. Jackson, “Homology-modelling protein-ligand interactions: Allowing for ligand-induced conformational change,” J. Mol. Biol., 2010.
- L. L. Porter and G. D. Rose, “Redrawing the Ramachandran plot after inclusion of hydrogen-bonding constraints,” Proc. Natl. Acad. Sci. U. S. A., 2011.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss, and J. M. Thornton, “PROCHECK: a program to check the stereochemical quality of protein structures,” J. Appl. Crystallogr., 1993.

