
Manuscript accepted on :
Published online on: 27-11-2015
Plagiarism Check: Yes
Azizeh Asadzadeh1*, Afshin Fassihi2, Parichehreh Yaghmaei1*, Morteza Pourfarzam3
1Department of Biology, Science and Research Branch, Islamic Azad University, Tehran, Iran 2Department of Medicinal Chemistry, School of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran 3Department of Biochemistry, School of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran. Corresponding Author Email : az.asadzadeh@yahoo.com
DOI : https://dx.doi.org/10.13005/bpj/796
Abstract
Tyrosinase (E.C, 1.14.18.1) catalyzes two key reactions in mammalian melanogenesis. Hyperpigmentation caused by the overproduction of melanin in the skin. Enzymatic browning of fruits and vegetables and derived products is caused by tyrosinase. Therefore, tyrosinase inhibitors have potential applications in medicine, cosmetics and agriculture. The aim of this research is the bioinformatical study of tyrosinase inhibition by some Kojic acid Derivatives. In order to investigating the mode of interaction of the compounds with tyrosinase active site, Chemical structures of all compounds were designed using ChemDraw program, then were transferred into Hyperchem software and energy minimized. docking study was performed by Auto Dock 4.2 program and The resulting docking poses were analyzed in Auto Dock Tools, DS Visualizer and Ligplot softwares. Among the all studied compounds, Ligands 12, 20, 21 and 23 displayed good docking results, In fact, these compounds have the most negative ΔGbind that indicated favorable interactions with the key amino acid residues at active site of tyrosinase. The docked conformation revealed that these compounds could form metal-ligand interaction with The Cu2+ ions in the active site. These insilico results can thus serve as a template for further studies invitro and invivo.
Keywords
binding energy; Docking; Kojic acid; Tyrosinase
Download this article as:| Copy the following to cite this article: Asadzadeh A, Fassihi A, Yaghmaei P, Pourfarzam M. Docking Studies of Some Novel Kojic Acid Derivatives As Possible Tyrosinase Inhibitors. Biomed Pharmacol J 2015;8(2) |
| Copy the following to cite this URL: Asadzadeh A, Fassihi A, Yaghmaei P, Pourfarzam M. Docking Studies of Some Novel Kojic Acid Derivatives As Possible Tyrosinase Inhibitors. Biomed Pharmacol J 2015;8(2). Available from: http://biomedpharmajournal.org/?p=1834 |
Introduction
Tyrosinase is copper-dependent enzyme that catalyze the oxidations of both monophenols and o-diphenols into reactive o-quinones (1-4). Melanin synthesis is initiated from tyrosine by tyrosinase. Although melanin is essential for protecting skin against UV irradiation damage, abnormal melanin production can lead to hyperpigmentation disorders such as melasma, lentigines, and post-inflammatory hyperpigmentation (5-7). Enzymatic browning of fruits and vegetables takes place in the presence of oxygen when tyrosinase and its polyphenolic substrates are mixed after brushing and crushing operations, which leads to the rupture of cell structure (8, 9). Browning in crops is unfavorable and decreases the commercial value of the products (2, 10). Furthermore, tyrosinase inhibitors have been used as insecticides and insect control agents for years, because this enzyme is associated with the host defense, wound healing, molting and sclerotisation of insects (11-14). Therefore, tyrosinase inhibition has become increasingly important for scientists in many branches of life science research.
The most intensively studied inhibitor of tyrosinase, 5-hydroxy-2-(hydroxymethyl)-4-pyrone, also termed as kojic acid (KA), was discovered in Japan by Saito in 1907 and is produced by various fungies (15). KA chelates transition metal ions such as Cu2+ and Fe3+ and is an effective free radicals scavenger. This inhibitor is currently applied as a food additive to prevent enzymatic browning of fruits. KA is also added to cosmeceuticals such as skin-lightening agents. It inhibits both the cresolase and catecholase activity of mushroom tyrosinase. However, its use has been limited, for its cytotoxicity, the skin irritation caused by its use in cosmetics and also its instability during storage (16, 17). Therefore, the development of novel, potent, non-toxic and stable kojic acid derivatives as tyrosinase inhibitors is of great importance.
Determination of the binding mode and affinity between the ligand and receptor is an essential issue in understanding the interaction mechanisms and designing therapeutic interventions in molecular recognition. The main aim of molecular docking is to accurately predict the conformation of a ligand locked up to the receptor binding site and to correctly estimate the strength of binding. Docking technique is a structure-based virtual screening that predicts the preferred binding orientation of the ligand to the desired receptor in a stable complex with respect to experiment (18). This technique searches over many possible interactions in order to identify a set of ligand poses that represent local minimum-energy positions of the ligand. It calculates a binding energy that can be used to accurately rank-order different ligands relative to experimentally measured binding affinities (19). this method is very informative to represent key structural characteristics and interactions to provide helpful data for proposing effective receptor inhibitors.
In the present study, twenty four derivatives of 3-hydroxypyridine-4-one scaffold were subjected to molecular docking studies. The preparation of some of these compounds is reported previously by this group (20, 21). These compounds are very close analogues of kojic acid having structural elements which provides them the ability to chelate copper ions and set up interactions with key residues located in the active site. Based on the in vitro evaluations some of these compounds had tyrosinase inhibition activity and were considered in the design of the study compounds (21).
Materials and Methods
For the protein–ligand docking simulation, we used AutoDock4.2 software package (22). Lamarckian Genetic Algorithm of the AutoDock 4.2 program was used to perform the flexible-ligand docking studies. An Intel-based core i5 personal computer with Linux (redhat 6) operating system was used for docking procedure. The crystal structure of tyrosinase from Agaricus bisporus (AbTYR; PDB code, 2Y9X) was chosen as the protein model for the present study.
Ligand Structure Preparation
General structure and the structural detailes of the compounds subjected to molecular docking simulations are provided in Table 1. All 2D structures of compounds were built using ChemDraw program (ChemDraw Ultra 10.0, Cambridge soft.), then were transferred into Hyperchem 8.0 software (HyperChem, Release 8.0 for Windows, Molecular Modeling System: HyperCube, 2007) and energy minimized with MM+ force field using Polak-Ribiere conjugate gradient algorithm until the root mean square gradient of 0.01 Kcal/Ǻ.mol. These optimized structures were used as inputs of the AutoDock tools. Then the partial charges of atoms were calculated using the Gasteiger-Marsili procedure implemented in the AutoDock tools package. Non-polar hydrogens of the compounds were merged and then rotatable bonds were assigned.
Protein Structure Preparation
A tyrosinase from the mushroom Agaricus bisporus has been successfully crystallized by Wangsa Ismaya and its structure has been elucidated by means of X-ray crystallography. The enzyme is commonly found as a H2L2 tetramer. The H subunit is the tyrosinase domain with a molecular mass of 43 kDa. This subunit contains two copper atoms coordinated by six histidine residues, His85, His61, His94, His259, His263 and His296. The L subunit is 14 kDa with a lectin-like fold and the function of this subunit is unknown. This tetramer is stabilized by two holmium ions in the H_H interface (8).
Co-crystallized ligands (tropolone), all chains except chain A, two holmium ions and water molecules of crystallization were removed from the complex using Discovery StudioVisualizer (23). All missing hydrogens were added and after determining the Kolman united atom charges, non-polar hydrogens were merged to their corresponding carbons using Autodock tools (24).
 |
Table 1: General structures and structural details of the studied compounds |
Docking Procedure
The AutoGrid program performs pre-calculations for the docking of a ligand to a set of grids that describe the effect that the protein has on point charges. The effect of these forces on the ligand is then analyzed by the AutoDock program. Using Autogrid as a part of the Auto dock package, desolvation parameters and electrostatic interactions were assigned to each protein atom. The grid points were set as 40 × 40 × 40 with the spacing valued at 0.375(a quarter of the C-C bond) to the catalytic site of the tyrosinase while the grid center was placed between the two metal ions located in the active site. The Lamarckian Genetic Algorithm (LGA) approach was selected as the search algorithm for the global optimum binding position search among the three different search algorithms offered by AutoDock 4.2. This algorithm has been determined superior compared with simulated annealing and genetic algorithm. 100 independent docking runs were carried out for each ligand. At the end of docking, the resulting docking poses were analyzed in AutoDockTools, DS Visualizer 3.5 and Ligplot softwares (25)
Results
Tropolone was redocked into the active site of tyrosinase for the method validation in the first step of docking procedure. The binding energy of the best bound conformation of tropolone was -3.11 kcal/mol. The molecular interactions of Tropolone with tyrosinase are illustrated in figure 1 and 2 . Tropolone interacts with HIS263 through π–π and cation-π interactions (orange lines) and its OH group forms a hydrogen-bond with Asn260 (black dashed line) (Figure 1). This Compound interacts hydrophobically with Gly281, His259, Val283, Ser282, His61, His263, Ala286 and Met280.
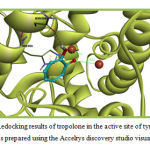 |
Figure 1: Redocking results of tropolone in the active site of tyrosinase. This figure was prepared using the Accelrys discovery studio visualizer program. |
After validation of the docking protocol, the 3D structures of the study compounds were docked into the tyrosinase active site. Docking analysis revealed that all active compounds, occupied the same space as tropolone with a similar binding mode. The estimated free binding energy values (ΔGbind) of the docked positions, intermolecular energy, electrostatic energy, total internal energy and torsional energy of these inhibitors into the active site are listed in Table 2. ΔGbind is the sum of the intermolecular energy and the torsional free-energy penalty and indicates favorable interactions and tight binding with key amino acid residues at the active site. Intermolecular energy is the sum of van der Waals, hydrogen binding, desolvation and electrostatic-energies (18, 19). The favorable interactions with the key amino acid residues at the active site of the enzyme are presented Tables 3.
Table 2: Docking results of kojic acid derivatives docked into tyrosinase active site. The values are expressed in kcal/mol
| Compound | Binding energy
|
Intermol energy | Electrostatic energy | Total internal
energy |
Torsional energy |
| 1 | -2.47 | -3.84 | -0.78 | -0.31 | 1.37 |
| 2 | -3.08 | -4.18 | -2.05 | -0.49 | 1.1 |
| 3 | -2.33 | -3.70 | -1.04 | -0.53 | 1.37 |
| 4 | -2.79 | -3.89 | -1.54 | -0.55 | 1.1 |
| 5 | -2.39 | -3.49 | -1.42 | -0.43 | 1.1 |
| 6 | -2.3 | -3.4 | -1.39 | -0.61 | 1.1 |
| 7 | -2.63 | -3.73 | -1.97 | -0.27 | 1.1 |
| 8 | -3.11 | -4.21 | -1.96 | -0.15 | 1.1 |
| 9 | -2.25 | -3.62 | -0.71 | -0.54 | 1.37 |
| 10 | -2.69 | -3.52 | -1.66 | 0. 46 | 0.82 |
| 11 | -3.51 | -4.88 | -1.74 | 0. 47 | 1.65 |
| 12 | -13.24 | -14.61 | -13.31 | -0.63 | 1.37 |
| 13 | -1.3 | -2.95 | 0.08 | 0.47 | 1.65 |
| 14 | -2.8 | -5.0 | -0.23 | -1.36 | 2.2 |
| 15 | -2.65 | -4.3 | +0.36 | -1.2 | 1.65 |
| 16 | -3.92 | -5.56 | -0.71 | -0.94 | 1.65 |
| 17 | -0.58 | -2.22 | +0.55 | -1.3 | 1.65 |
| 18 | -2.13 | -3.78 | +0.01 | -1.15 | 1.65 |
| 19 | -2.85 | -3.94 | -1.91 | 0.23 | 1.1 |
| 20 | -12.23 | -13.6 | -13.35 | -0.34 | 1.37 |
| 21 | -11.87 | -14.07 | -14.41 | -1.02 | 2.2 |
| 22 | -2.03 | -4.5 | -1.32 | -1.86 | 2.47 |
| 23 | -12.21 | -14.4 | -13.43 | -0.02 | 2.2 |
| 24 | -2.2 | -4.67 | -0.89 | -0.76 | 2.47 |
| Tropolone | -3.11 | -3.38 | -1.51 | -0.03 | 0.27 |
Table 3: Interactions between the study compounds and tyrosinase active site residues. The value in parenthesis is the distance between donor atom and Cu2+ ion expressed in angstrom (Ǻ)
|
Compound |
Interaction with Cu+2
|
Interaction with amino acid residues
|
||
| H-bonding | Pi interactions | Hydrophobic | ||
| 1 | C=O… Cu2+ (2.521) | His263
|
His263
(sigma- π) |
Gly281, Met280, Asn260, His85, His244, Phe264, Val283, Ser282, His61, His259, His263, Ala286 |
| 2 | C=O… Cu2+ (2.130) | His 61, His296 | – | Gly281, Met280, His85, Val283, Ser282, His259, His263, Phe264, Ala286, Phe292, Phe90 |
| 3 | C=O… Cu2+ (2.430) |
Asn260 |
His263
(sigma- π) |
His85, His244, Val283,His61,His259, His263, Phe292, Ala286, Glu322 |
| 4 | C=O… Cu2+ (2.367) |
His 263 |
His263
(sigma- π) |
Gly281, Val283, Ser282, His61, His259, His263, Phe264 ,Ala286, Met280 |
| 5 | C=O… Cu2+ (2.523) |
His 263 |
His263
(sigma- π) |
Gly281, Asn260, His85, Val283, Ser282, His61, His259, His263, Phe264, Ala286 |
| 6 | C=O… Cu2+ (2.523) |
His 263, His 259 |
– | Gly281, Asn260, His85, His244,Val283,Ser282, His61, His259, His263, Phe264, Ala286 |
| 7 | C=O… Cu2+ (2.348)
C=O… Cu2+ (2.324) |
Asn260 |
– | Gly281, His85, Val283, His259, His263, Phe264, Ala286, Phe292 |
| 8 | C=O… Cu2+ (2.450)
C=O… Cu2+ (2.333) |
Asn260, His259, His296 |
– | Gly281, His85, Val283,Ser282, His61, His263, Phe264, Ala286, Phe292 |
| 9 | C=O… Cu2+ (2.213) |
Asn260
|
– | Gly281, His85, Val283,Ser282, His61, His263, Phe264, Ala286, Phe292 |
|
10 |
C=O… Cu2+ (2.344) |
His263, His61 |
– | Gly281, Met280, Asn260, Val283,Ser282, His61, His259, His263, Phe264, Phe292 |
| 11 | – | His61,His85,Val283,Gly281 | – | Gly281, Asn260, Ser282,His259, His263, Ala286 , Phe292 |
| 12 | N=O… Cu2+ (2.255)
N=O… Cu2+ (2.279) |
His259, Asn260 His296, His61 His244, Glu256 |
– | Gly281, His94, Val283, Ser282, His61, His263, Asn260, His259, Phe264, Phe90, Phe292, Ala286
|
| 13 | – | Gly281, Met280 | – | Val283, Ser282, His61,His259, His263, Phe264, Phe292, His263 |
| 14 | – | Gly281, Met280, Asn260, Asn260 | – | His85, Val283, Ser282, His61, His259, Pro277, Pro284, His263, Phe264, Ala286, Phe292 |
| 15 | – | Gly281, Met280, Asn260, Asn260 | Phe264
(sigma- π) |
His85, Val283, Ser282, His61, His259, Pro277, His263, Phe264, Ala286,, Phe292, Arg268 |
| 16 | – | Asn81, His85, Val283, Ser282,
Cys83 |
– | Pro284, His263, Phe264, Ala286,, Phe292, Asn260, Asn81, Thr324, Thr84, Cys83 |
| 17 | – | Gly281, Met280, Asn260, Asn260 | – | Val283, Ser282, His61,His259, Ala286, Pro284, Gly281, His263, Phe264, Phe292
|
|
18 |
– | Gly281, Met280, Asn260, Asn260 | – | Val283, Ser282, His85,His259, Ala286, Pro284, His263, Phe264, Phe292, His85, His85 |
| 19
|
– | His296, His61, Asn260 | – | Val283, Ser282, His85,His259, Ala286, Gly281, His263, Phe264, Phe292,, Phe90 |
| 20 | C=O… Cu2+ (2.124)
C-O… Cu2+ (2.097) |
His85, Ser282 | His263
(sigma- π) |
Asn260, Val283, Ser282, His61, His259, Phe292, Ala286 |
| 21
|
C=O… Cu2+ (1.980)
C-O… Cu2+ (2.174) |
His244, His296, His259, Glu256,
Asn260 |
– | Asn260, Val283, His263, Phe264, Val248 |
| 22 | – | Val283, His263 | – | Gly281, Asn260, His85, His244, Ser282, Glu256, His259, His263, Phe264 |
| 23 | C=O… Cu2+ (2.373)
C-O… Cu2+ (2.096) |
His296, His259
|
– | Met280, Asn260, Val283, Ser282, His61, His259, His94, Phe90, Phe264, Phe292 |
| 24 | – | His244, His244, His263, Asn260, Asn260 | – | Met280, Val283, Ser282, His85,His263, Gly281, Phe264, |
| Tropolone | C=O… Cu2+ (2.519) |
Asn260 |
His263
(cation-π) (π -π) |
Gly281, His259, Val283, Ser282, His61, His263, Ala286, Met280 |
Disscussion
The binding mode of the study compounds against tyrosinase was investigated by performing molecular docking smulations. There is no X-ray crystallography data available for the three-dimensional structure of human tyrosinase (26). Thus, the 3D structure of Agaricus bisporus mushroom tyrosinase was used for the docking study. Possible H-bonding, π-sigma, cation-π and metal interactions assessed using the Discovery StudioVisualizer program. Ligplot software predicted the possible hydrophobic interactions between the docked ligands and the catalytic site residues.
Based on the structural diversity of the compounds, they are divided into three different groups. Interactions of the most active compounds in each group are provided in related sections.
Group I: Compounds 1-12
Compound 12 was the most active compound in this group. This compound interacts through H-bonding with The His61, His259, His296, His244, Asn260 and Glu256 residues of tyrosinase active site. The ΔGbind for this molecule was -13.24kcal/mol. Compound 12 interacts hydrophobically with Gly281, His94, Val283, Ser282, His61, His263, Asn260, His259, Phe264, Phe90, Phe292 and Ala286. The docking results for the most active compound in this group, are illustrated in Figure 2. The docked conformation revealed that the NO2 group of compound 12, which resided 2.25-2.27Å adjacent to the di-copper nucleus, could form metal-ligand interaction.
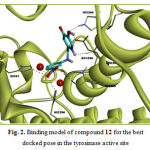 |
Figure 2: Binding model of compound 12 for the best docked pose in the tyrosinase active site |
Group II: Compounds 13-19
Molecular docking study revealed that compounds 13-19 may attach to tyrosinase active site via H-bond and hydrophobic interactions as other compounds. Members of this group exhibited weaker affinity to tyrosinase compared with the previous group. Compound 17 in this group exhibited the weakest affinity amongst the studiy compounds. None of the members in this group had interaction with the di-copper ion in the active site. The ΔGbind of the most active compound, Compound 16, in this group was -3.92 kcal/mol. Asn81, His85, Val283, Ser282 and Cys83 of tyrosinase were the sites for hydrogen bonding interactions with compound 16. This Compound interacts hydrophobically with Pro284, His263, Phe264, Ala286, Phe292, Asn260, Asn81, Thr324, Thr84 and Cys83. Some of these interactions are not common with Co-crystallized ligands (tropolone). The docking results for compound 16 are illustrated in Figure 3.
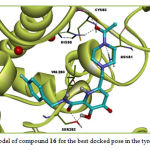 |
Figure 3: Binding model of compound 16 for the best docked pose in the tyrosinase active site |
Group III: Compounds 20-24
Compounds 22 and 24 exhibited the lowest interactions with tyrosinase in this group. They both did not interact with the di-copper ion in the active site. They lacked a free diketo acid group and due to the improper orientation of the esterified diketo acid moiety, were not able to have any interaction with the metal ions. Ligands 20, 21 and 23 displayed good docking results, with 20 as the most potent member of the group (-12.23 kcal/mol). In the case of compound 20, the carboxylic acid group substituted on C2 and the α– hydroxy ketone moiety on the 3-hydroxypyridin 4-one ring together may form metal-ligand interactions with two metal ions as enolyzed diketo may in compounds 21 and 23. Two carbonyl moieties of compound 20 formed two hydrogen bonds with His85 and Ser282 and interacted again with the imidazole ring of His263 by a sigma–π interaction. These observations are illustrated in Figure 4. This compound makes hydrophobic interactions with Asn260, Val283, Ser282, His61,His259, Phe292 and Ala286. In compounds 21 and 23 the diketo acid moiety may interact with both metal ions in the active site. It is demonstrated with molecular docking results (Table 3).
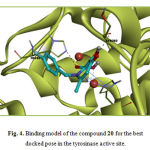 |
Figure 4: Binding model of the compound 20 for the best docked pose in the tyrosinase active site. |
Conclusion
In agreement with binding energies, Ligands 12, 20, 21, and 23 displayed good docking results. The docking results for this compound are in accordance with the docking results reported by others in terms of the amino residues involved in interaction with the inhibitor molecule (1, 26-29).
All of these four compounds have an efficient metal-ligand interaction with the Cu2+ ions. In tyrosinase enzyme, each copper ion is coordinated by three histidine residues (4). Among these six histidines, His61, His296 and His259 interact with compound 12, His58 interact with compound 20, His61, His296 and His259 interact with compound 21 and His296 and His259 interact with compound 23. Among the all studied compounds, the best docking result was obtained for compound 12. In fact, this compound had the most negative ΔGbind (-13.24Kcal/mol) that indicated favorable interactions and tight binding with the key amino acid residues at active site of tyrosinase. These insilico results can thus serve as a template for further studies invitro and invivo.
Acknowledgment
The author would like to thank Islamic Azad University for providing research facilities.
References
- Chung KW, Jeong HO, Jang EJ, Choi YJ, Kim DH, Kim SR, et al. Characterization of a small molecule inhibitor of melanogenesis that inhibits tyrosinase activity and scavenges nitric oxide (NO). Biochimica et Biophysica Acta (BBA)-General Subjects. 2013;1830(10):4752-61.
- de Faria RO, Moure VR, de Almeida Amazonas ML, Krieger N, Mitchell DA. The biotechnological potential of mushroom tyrosinases. Food Technology and Biotechnology. 2007;45(3):287.
- Haghbeen K, Tan EW. Direct spectrophotometric assay of monooxygenase and oxidase activities of mushroom tyrosinase in the presence of synthetic and natural substrates. Analytical biochemistry. 2003;312(1):23-32.
- Ismaya WT, Rozeboom HJ, Weijn A, Mes JJ, Fusetti F, Wichers HJ, et al. Crystal structure of Agaricus bisporus mushroom tyrosinase: identity of the tetramer subunits and interaction with tropolone. Biochemistry. 2011;50(24):5477-86.
- Agar N, Young AR. Melanogenesis: a photoprotective response to DNA damage? Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis. 2005;571(1):121-32.
- Hamilton A, Gomez B. Melanins in fungal pathogens. Journal of medical microbiology. 2002;51(3):189.
- TH Khan M. Novel tyrosinase inhibitors from natural resources–their computational studies. Current medicinal chemistry. 2012;19(14):2262-72.
- Chang T-S. An updated review of tyrosinase inhibitors. International journal of molecular sciences. 2009;10(6):2440-75.
- Muñoz-Muñoz JL, del Mar García-Molina M, Garcia-Molina F, Berna J, Garcia-Ruiz PA, García-Moreno M, et al. Catalysis and inactivation of tyrosinase in its action on o-diphenols, o-aminophenols and o-phenylendiamines: potential use in industrial applications. Journal of Molecular Catalysis B: Enzymatic. 2013;91:17-24.
- Loizzo M, Tundis R, Menichini F. Natural and synthetic tyrosinase inhibitors as antibrowning agents: an update. Comprehensive Reviews in Food Science and Food Safety. 2012;11(4):378-98.
- Beerntsen BT, James AA, Christensen BM. Genetics of mosquito vector competence. Microbiology and Molecular Biology Reviews. 2000;64(1):115-37.
- Chai W-M, Liu X, Hu Y-H, Feng H-L, Jia Y-L, Guo Y-J, et al. Antityrosinase and antimicrobial activities of furfuryl alcohol, furfural and furoic acid. International journal of biological macromolecules. 2013;57:151-5.
- Guerrero A, Rosell G. Biorational approaches for insect control by enzymatic inhibition. Current medicinal chemistry. 2005;12(4):461-9.
- Li Z-C, Chen L-H, Yu X-J, Hu Y-H, Song K-K, Zhou X-W, et al. Inhibition kinetics of chlorobenzaldehyde thiosemicarbazones on mushroom tyrosinase. Journal of agricultural and food chemistry. 2010;58(23):12537-40.
- Aytemir MD, Karakaya G, Ekinci D. Kojic acid derivatives: INTECH Open Access Publisher; 2012.
- Burdock GA, Soni MG, Carabin IG. Evaluation of health aspects of kojic acid in food. Regulatory toxicology and pharmacology. 2001;33(1):80-101.
- Rho HS, Lee CS, Ahn SM, Hong YD, Shin SS, Park Y-H, et al. Studies on tyrosinase inhibitory and antioxidant activities of benzoic acid derivatives containing kojic acid moiety. Bulletin of the Korean Chemical Society. 2011;32(12):4411-4.
- Huang S-Y, Zou X. Advances and challenges in protein-ligand docking. International journal of molecular sciences. 2010;11(8):3016-34.
- Mukesh B, Rakesh K. Molecular docking: a review. Int J Res Ayurveda Pharm. 2011:1746-51.
- Mohammadpour M, Sadeghi A, Fassihi A, Saghaei L, Movahedian A, Rostami M. Synthesis and antioxidant evaluation of some novel ortho-hydroxypyridine-4-one iron chelators. Research in pharmaceutical sciences. 2012;7(3):171.
- Saghaie L, Pourfarzam M, Fassihi A, Sartippour B. Synthesis and tyrosinase inhibitory properties of some novel derivatives of kojic acid. Research in pharmaceutical sciences. 2013;8(4):233.
- Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. Journal of computational chemistry. 2009;30(16):2785-91.
- Visualizer DS. Release 3.5. Accelrys Inc, San Diego, CA, USA. 2012.
- Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, et al. A new force field for molecular mechanical simulation of nucleic acids and proteins. Journal of the American Chemical Society. 1984;106(3):765-84.
- Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein engineering. 1995;8(2):127-34.
- Wen K-C, Chang C-S, Chien Y-C, Wang H-W, Wu W-C, Wu C-S, et al. Tyrosol and its analogues inhibit alpha-melanocyte-stimulating hormone induced melanogenesis. International journal of molecular sciences. 2013;14(12):23420-40.
- Vontzalidou A, Zoidis G, Chaita E, Makropoulou M, Aligiannis N, Lambrinidis G, et al. Design, synthesis and molecular simulation studies of dihydrostilbene derivatives as potent tyrosinase inhibitors. Bioorganic & medicinal chemistry letters. 2012;22(17):5523-6.
- Wang Z-J, Lee J, Si Y-X, Oh S, Yang J-M, Shen D, et al. Toward the inhibitory effect of acetylsalicylic acid on tyrosinase: Integrating kinetics studies and computational simulations. Process Biochemistry. 2013;48(2):260-6.
- Pei C-J, Lee J, Si Y-X, Oh S, Xu W-A, Yin S-J, et al. Inhibition of tyrosinase by gastrodin: An integrated kinetic-computational simulation analysis. Process Biochemistry. 2013;48(1):162-8.

