
Manuscript accepted on :01-Oct-2018
Published online on: 09-10-2018
Plagiarism Check: Yes
Reviewed by: Souhail Regragui
Second Review by: Bibhas Kar
Final Approval by: Dr Ayush Dogra
I. B. Putra Adnyana , T. G. N. Chandragiram and Ketut Surya Negar
, T. G. N. Chandragiram and Ketut Surya Negar
Department of Obstetrics and Gynecology, Faculty of Medicine, Udayana University/Sanglah General Hospital, Denpasar-Bali.
Corresponding Author E-mail: tutsuryanegara@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/1584
Abstract
Bilateral laparoscopic gonadectomy is reported as an operative procedure of removing the intraabdominal gonads in individuals with complete androgen insensitivity syndrome (CAIS). We present a case of 18 year old patient who had history of primary amenorrhea. The clinical evaluation shows a female phenotype, excellent breast development, absence of hair in the groins and axillary areas and short blind vagina. In ultrasonographic examination uterus was absent. Hormonal assay showed elevated serum testosterone levels of 1,037 ng/dL. A karyotype was also performed which revealed a 46 XY. Laparoscopy diagnostic revealed absence of internal genitalia except bilateral gonads appearing as testes. The testes were removed to avoid the risk of malignancy. Histopathological examination revealed that both the masses were composed of testicular tissue. Laparoscopy allows to clearly identify, locate as well as gonadectomy in the same time. This approach results in rapid recovery with minimal blood loss and should be considered for all patient with intraabdominal gonads.
Keywords
CAIS; Gonadectomy; Laparoscopy
Download this article as:| Copy the following to cite this article: Adnyana I. B. P, Chandragiram T. G. N, Negara K. S. Laparoscopy Diagnostic and Bilateral Gonadectomy in a Patient with Complete Androgen Insensitivity Syndrome (CAIS) – A Rare Case of Primary Amenorrhoea with 46XY karyotype. Biomed Pharmacol J 2018;11(4). |
| Copy the following to cite this URL: Adnyana I. B. P, Chandragiram T. G. N, Negara K. S. Laparoscopy Diagnostic and Bilateral Gonadectomy in a Patient with Complete Androgen Insensitivity Syndrome (CAIS) – A Rare Case of Primary Amenorrhoea with 46XY karyotype. Biomed Pharmacol J 2018;11(4). Available from: http://biomedpharmajournal.org/?p=23543 |
Introduction
Androgen insensitivity syndrome (AIS) or testicular feminization syndrome (TFS) is the most common diagnosis of a rare inherited form of male pseudohermaphroditism. Based on karyotyping the patient usually has a normal 46 XY but present as a female phenotype, typical clinical presentation of primary amenorrhea in an adolescent and impaired development of secondary sexual characteristics, presence of nondysplastic testis, undeveloped of uterus, fallopian tubes, cervix and a vagina of variable length.1,2,3 In general population, this syndrome affect at least 1 in 20,000 births and the most common form of this syndrome is a complete form (CAIS) which occurs 1 in 20,000 to 64,000 male births.4 As high as 30% the nondysplastic testis could be progess to malignancies after puberty period which is commonly as a malignant dysgerminoma, therefore early evaluation and gonadectomy is mandatory.1,5,6,7
A few cases of laparoscopic gonadectomy have been reported allowing at the same time of both identification and removal of the gonads. In this report we present our experience in the laparoscopic approach in CAIS.
Case
A 18 year old unmarried young phenotypic female came with primary amenorrhea and was evaluated through a diagnostic procedure that included clinical examination, hormonal assay and karyotyping. The BMI was 23.57 kg/m2, height 157 cm and weight 58 kg. Physical examination revealed a female phenotype with normal skeletal proportion, normal development of breast (Tanner stage-5) but with absence of hair in the groins and axillary area. The vulva and perineum appeared normal, vagina was present 3 cm in length but ended blindly. The transabdominal pelvic ultrasonography confirmed the absence of uterus and ovaries. Hormonal assay showed elevated serum testosterone levels of 1,037 ng/dL. A karyotyping was also done which was 46 XY. In view of these findings, a diagnosis of CAIS was made. The patient was counselled regarding the need for gonadectomy in view of increased risk of malignancy and also regarding the need for lifelong hormone replacement therapy. Diagnostic and operative laparoscopy was performed under general anesthesia after standard preoperative preparation. Pelvic inspection revealed absent of internal genitalia except, bilateral gonads inside the abdominal cavity appearing as normal size testes attached at prepsoic region (Figure 1 and 2). They were subsequently dissected and removed. The specimen sent for histopathological examination. Postoperative recovery was uneventful, the patient was discharged after one day.
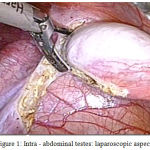 |
Figure 1: Intra – abdominal testes: laparoscopic aspect.
|
Excision of Right gonad and spermatic cord done by laparascopy.
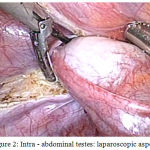 |
Figure 2: Intra – abdominal testes: laparoscopic aspect.
|
Excision of Left gonad and spermatic cord done by laparascopy.
Gross examination of the gonadectomy specimen revealed a smooth, glistening mass with a solid grey-brown cut surface. The histopathologic report revealed seminiferous tubules lined predominantly by sertoli cells with peritubular fibrosis. No ovarian tissue was seen. The interstitium showed Leydig cell hyperplasia. No signs of malignancy were identified in both the gonads (Figure 3). The patient was put on oestrogen therapy and informed that she could have a normal sexual life but would be infertile.
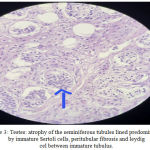 |
Figure 3: Testes: atrophy of the seminiferous tubules lined predominantly by immature Sertoli cells, peritubular fibrosis and leydig cel between immature tubulus.
|
Histopathological aspect, HE staining: ob.10x
Discussion
Androgen insensitivity syndrome (AIS) or testicular feminization syndrome (TFS), is a rare disease when a male, genetically XY, because of various abnormalities of the X chromosome become resistant to the actions of androgen hormones, has some physical characteristic of a female, or even a full female phenotype.8 AIS is the most frequent X-linked recessive disorders of sex development and the most common type of male pseudohermafroditism. It characterized by complete or partial inability of cell respond to androgen secondary by mutation in the androgen receptor gene located on long arm of the X chromosome (locus Xq11-12) in the form of complete or partial deletions, point mutations or small insertions. Although 70% of these mutations are X-linked recessive, 30% can also be sporadic de novo mutations.9,10
According to the severity of androgen resistence, AIS represents a spectrum of phenotypic disorders that varies from pure female complete androgen insensitivity syndrome (CAIS), ambiguous genitalia partial androgen insensitivity (PAIS) and to phenotypically infertile male minimal androgen insensitivity form (MAIS). The complete form is 10 times more common than the incomplete form. Most of cases are diagnosed in the newborn period with inguinal masses. But others also present in late adolescent by the presence of primary amenorrhoea.8,11,12
AIS is diagnosed based on the following findings: absence of female internal genital organs on physical exam and pelvic ultrasonography, karyotyping, molecular genetic testing of the AR gene mutations (chromosomal locus Xq11-q12), and elevated testosterone and luteinizing hormone levels.13,14 In our case the diagnosis of AIS was based on gynecologic examination, pelvic ultrasound, hormonal analysis, diagnostic laparoscopy findings, karyotyping and histopathological study. The initial presentation in our case was primary amenorrhea, with a full female phenotype but without pubic and axillary hair, absence of female internal genital organs on physical exam, pelvic ultrasonography and laparoscopy, karyotyping which was 46 XY.
Elevated serum levels of testosterone and of LH, as endocrine features of CAIS at puberty, due to the androgen insensitivity and the consequent lack of negative feedback exerted by sex hormone on hypothalamus and hypophysis. Testosterone itself becomes target of aromatase, and is converted to estrogens. For this condition CAIS patients present with higher estrogens levels than normal male and have excellent development of breast as well as the present case. Testosterone level had also elevated in our patient.11,12 Anti-Müllerian Hormone (AMH) concentration in patients with AIS is normal, as the secretion and function of sertoli cell is not impaired. The testes continue to develop but usually remain either in the abdomen (21%), the groin (60%), or labia majora. Testosterone is formed normally during intrauterine life but due to inability of the target organs to respond to it, male characteristics fail to develop and the result is somatic female without any mullerian structures, such as uterus or fallopian tubes since the posterior vagina is also of mullerian origin its development, too, is inhibited and short blind vagina results. Pelvic ultrasonography usually shows absence of mullerian derivates and vaginal examination reveals a blind-ending vagina without a cervix. Our patient had all these features.11,15,16
Treatment of CAIS includes removal of the testes after puberty, when feminization is complete, in order to prevent testicular malignancy. Laparoscopy allows to clearly identify, locate as well as remove the gonads through a small incision. Localization of gonads is easier by laparoscopy as compared to laparotomy, due to the better visualization of the entire abdomen and pelvis.17,18,19 Laparoscopic removal of gonads has many advantages: minimal blood loss, rapid recovery, shorter hospital stays and minimum psychological trauma compared to laparotomy. The operational time is similar in laparoscopy and laparotomy.18,20
Conclusions
Bilateral laparoscopic gonadectomy is a minimally invasive effective procedure of removing the intraabdominal gonads in individuals with CAIS. This approach results in rapid recovery with minimal blood loss and should be considered for all patient with intraabdominal gonads.
Conflict of Interest
The authors declares that there is no conflict of interest regarding the publication of this paper.
References
- Creatsas G. K. Hermaphroditism-Intersexual Disorders. Modern Obstetrics and Gynecology. Athens: Pashalidis Publishers. 1998;34-42.
- Speroff L., Glass R., Kase N., (eds). Normal and abnormal sexual development in Clinical Gynecologic Endocrinology and Infertility (5th) Williams and Wilkins Publishers. 1994;321
- Collins G. M., Kim D. U., Logrono R., et al. Pure seminoma arising in androgen insensitivity syndrome (testicular feminization syndrome) a case report and review of literature. Mod Pathol 1993;6(1):89-93.
- Barthold J. S., Kumasi-Rivers K., Upadhyay J., et al. Testicular position in the androgen insensitivity syndrome: implications for the role of androgens in testicular descent. J Urol 2000;164(2):497-501.
CrossRef - Griffin J. E., Wilson J. D. The syndromes of androgen resistance. N England J Med. 1980;302:198.
CrossRef - Diaz D. G., Chavez-Lopez D. Laparoscopic gonadectomy in a woman with complete androgen insensitivity syndrome. J Amer Assoc Gynec Laparosc. 1996;3:317.
CrossRef - Obianwu C. W., Palter S. F., Bruno A. A Jr. Bilateral laparoscopic gonadectomy in a patient with complete androgen insensitivity. A case report. J Reprod Med. 1996;15. 41:270.
- National Center for Biotechnology Information, US National Library of Medicine 2007; Available at http://www.ncbi.nlm.nih.gov/books/NBK1429/. (Accessed May 10, 2018).
- Sharma G., Ghode R. Androgen insensitivity syndrome: discussion based on three cases. Int J Reprod Contracept Obstet Gynecol. 2015;4(5):1585-1588.
CrossRef - Constantin G., Alexandru D., Sorin P., et al. Testicular feminization: complete androgen insensitivity syndrome. Discussions based on a case report. Rom J MorpholEmbryol. 2014;55(1):177-81.
- Kaur N., Goyal N., Singh H., et al. Testicular Feminization Syndrome- A Rare Case Report. Ann. Int. Med. Den. Res. 2017;3(5):01-03.
- Tarafdar R. L., Ahmed S. S. Androgen Insensitivity Syndrome- A Case Report. American Journal of Clinical and Experimental Medicine. 2015;4(3):133-136.
- Solari A., Groisman B., Bidondo M. P., et al. Complete androgen insensitivity syndrome diagnosis and clinical characteristics. Arch Argent Pediatr. 2008;106(3):265-268.
- Ahmed S. F., Cheng A., Dovey L., et al. Phenotypic features androgen receptor binding and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome. J Clin Endocrinol Metab. 2000;85:658-665.
CrossRef - Campo S., Stivel M., Nicolau G. Testicular function in post pubertal male pseudohermaphroditism. Clinical Endocrinology. 1979;11(5):481–490.
CrossRef - Galani A., Kitsiou-Tzeli S., Sofokleous C., et al. Androgen insensitivity syndrome clinical features and molecular defects. Hormones. 2008;7(3):217–229.
CrossRef - Tsugaya M., Hayashi Y., Mogami T., et al. Laparoscopic gonadectomy for testicular feminization syndrome. Int J Urol . 1995;2(3):200-202.
CrossRef - Kallipolitis G. K., Milingos S. D., Creatsas G. K., et al. Laparoscopic gonadectomy in a patient with testicular feminization syndrome. J Pediatr Adolesc Gynecol. 2000;13(1):23-26.
CrossRef - Kriplani A., Abbi M., Ammini A. C., et al. Laparoscopic gonadectomy in male pseudohermaphrodites. Eur J Obstet Gynecol Reprod Biol. 1998;81(1):37-41.
CrossRef - Chantilis S. J., Mc Quitty D. A., Priminger G. M., et al. Laparoscopic removal of gonads containing an occult seminoma in a woman with complete androgen resistance. J Am Assoc Gynecol Laparosc. 1994;1(3):277- 282.
CrossRef

