
Manuscript accepted on :13-Sep-2018
Published online on: 18-09-2018
Plagiarism Check: Yes
Reviewed by: Ahmar Rauf
Second Review by: Guy-Armel Bounda
Final Approval by: Alessandro Leite Cavalcanti
Ani R1, Roshini Manohar1, Gayathri Anil1 and O.S. Deepa2
1Department of Computer Science and Applications, Amrita Vishwa Vidyapeetham, Amritapuri, India.
2Department of Mathematics, Amrita Vishwa Vidyapeetham, Coimbatore, India.
Corresponding Author E-mail: anir@am.amrita.edu
DOI : https://dx.doi.org/10.13005/bpj/1518
Abstract
In earlier years, the Drug discovery process took years to identify and process a Drug. It takes a normal of 12 years for a Drug to travel from the research lab to the patient. With the introduction of Machine Learning in Drug discovery, the whole process turned out to be simple. The utilization of computational tools in the early stages of Drug development has expanded in recent decades. A computational procedure carried out in Drug discovery process is Virtual Screening (VS). VS are used to identify the compounds which can bind to a Drug target. The preliminary process before analyzing the bonding of ligand and drug protein target is the prediction of drug likeness of compounds. The main objective of this study is to predict Drug likeness properties of Drug compounds based on molecular descriptor information using Tree based ensembles. In this study, many classification algorithms are analyzed and the accuracy for the prediction of drug likeness is calculated. The study shows that accuracy of rotation forest outperforms the accuracy of other classification algorithms in the prediction of drug likeness of chemical compounds. The measured accuracies of the Rotation Forest, Random Forest, Support Vector Machines, KNN, Decision Tree and Naïve Bayes are 98%, 97%, 94.8%, 92.8%, 91.4%, 89.5% respectively.
Keywords
Classification Algorithms; Drug likeness; Ensemble Methods; Lipinski’s Rule; Molecular Descriptors; Virtual Screening
Download this article as:| Copy the following to cite this article: Ani R, Manohar R, Anil G, Deepa O. S. Virtual Screening of Drug Likeness using Tree Based Ensemble Classifier. Biomed Pharmacol J 2018;11(3). |
| Copy the following to cite this URL: Ani R, Manohar R, Anil G, Deepa O. S. Virtual Screening of Drug Likeness using Tree Based Ensemble Classifier. Biomed Pharmacol J 2018;11(3). Available from: http://biomedpharmajournal.org/?p=22699 |
Introduction
Drug discovery is the process of identifying potential drug for specified disease. The whole process takes many years. With the introduction of machine learning, the drug discovery process became easier. The fundamental objective of virtual screening is the contraction of the excessive virtual compound space of limited biological molecules. Virtual Screening utilizes computer based strategies to find new ligands on the base of biological structures. Initial stage of Virtual Screening is the evaluation of Drug likeness of small molecule. Drug likeness is defined as whether an existing molecule has the chemical properties similar to known drugs. The Drug likeness of chemical compounds considers Lipinski’s Rule as main criteria. It is an important rule to determine Drug likeness or decide if a chemical compound with certain molecular descriptors would make it an orally effective Drug in humans. The rule is significant during Drug discovery process. The rule depicts properties critical for a Drug’s pharmacokinetics in the human body, including their absorption, distribution, metabolism, excretion and toxicity. In most of the cases, of predicting Drug likeness the rules considered are Lipinski’s Rule of five and ADMET properties. This study consider eight different rules such as Lipinski’s rule of five, Ghose filter rule, Verber’s rule, CMC-50 rule, MDDR rule, BBB Likeness, Weighted QED and UnWeighted QED. The dataset consists of molecular descriptors of chemical compounds and also consists of the status of satisfying the above specified rules. The dataset focuses on chemical compounds available in medicinal plants and their molecular descriptors.
Support Vector Machines1 (SVM) proved to be very good classifier for classification problem. It is used for classification or regression. In n-dimensional space every data point is sketched in the case of SVM. There were limitations in SVM. Random Forest2 algorithm is an ensemble classifier based on the concept of bagging. It is used for feature engineering, which implies recognizing most essential feature out of available features from training dataset. Decision tree is another algorithm used for classification. It is generally illustrated as a tree, with the root at the top and leaves at the bottom. DT’s are mostly represented in top-down approaches. Another tree based ensemble method has been introduced which is Rotation forest. A recently recommended method for building classifier ensemble is Rotation Forest,3 which uses PCA, based transformation in subsets of data before applying decision tree. The current work is based on the study of molecular descriptors for Drug/ Non-Drug compounds extracted from medicinal plants. The molecular descriptors of these chemical compounds are identified. The machine learning approaches like classification of these compounds to drug compounds and non drug compounds are done. A comparison of different classification algorithms in the prediction of drug likeness of chemical compounds are carried out in this study.
The molecular descriptors are used for the prediction of drug likeness. In the case of data collection, the compounds of medicinal plants are considered. Data are collected based on eight rules such as Lipinski’s rule, Ghose filter rule, Verber’s rule, CMC-50 rule, MDDR rule, BBB Likeness, Weighted QED, UnWeighted QED. Initial study of this work begins with paper4 done by Ani R at el. In this work4 study and analysis of descriptors of 11compounds was done. The methods used in this paper were Random Forest, Decision Tree, Naïve Bayes and Rotation Forest. We extended the study with more compounds nearly 600 and extended with Kernel based methods (SVM) and nearest neighbor methods. In the present study, we have implemented Random Forest algorithm, K-Nearest Neighbor algorithm, and Decision tree, Naïve Bayes, SVM and Rotation Forest. A comparison between the accuracy of the algorithms is done.
Related Work
The study of SVM classifier in the classification of Drug and non-Drug compounds are detailed in paper.1 They used SVM with various Feature Selection approaches. In the preprocessing step, the number of features was reduced based on correlation method. Then SVM is applied to the training set. Further Feature Selection methods are used. It is vital to dispense potentially unwanted compounds such as passive or noxious particle. Some of the Feature Selection methods used in1 are SVM Recursive Feature Elimination, Wrapper method and Subset Selection. The chemical compounds for training and test set have been taken from.4 The goal is to design a hyper plane that classifies all training set into two classes. SVM with Subset Selection outrun better than logistic regressions, which was used in their previous study. The main drawback they specified is comparatively limited sample set. Secondly drawback is associated to training set that were totally unbalanced. Since these limitations are there large data set are needed to ratify the conclusion in future works.
In the study of Cano, Gaspar, et al.2 They have proven the power of automatic selection of characteristics using Random Forest. This paper covers the challenges of feature selection through computational intelligence methods. An appropriate selection of the set of molecular descriptors is basic to enhance the prediction and automatic selection of these descriptors. In this paper Random Forest is applied as a Feature selector. This paper used public datasets for analysis of classification conduct of the method. The main contribution of the paper is the automatic selection of a ranked and reduced subset of features to feed the classifier. Random Forest is used for both classification and regression. It’s used for feature engineering. It selects automatically the molecular descriptors which permit to enhance the integrity of the fitting process. Random forest is used for two purposes, one is feature ranking dimensionality reduction and other is classification using automatically selected feature subset. RF improves accuracy and reduces cost. In RF, a ranking of the contribution of each variable is determined to predict the output variable. The use of Random Forest not only improves the accuracy of the classification but also reduces the computational cost.2 The model behavior is influenced by two parameters: the number of trees and the number of partitions to be made (split). RF results outperform classification results provided by SVM. Future work include the automation of the choice of a learning algorithm depending of the characteristics of a given prediction, data source and prediction performance.
Methodology
Bagging is a machine learning algorithm which is used to shorten the variance. Bootstrap procedure develops various datasets and these bootstrap data are trained using classification algorithms. It is a method for generating various form of classifier and use this to get a combined classifier. Each base classifier is generated by different bootstrap samples. In a classification tree, bagging5 takes a majority vote from classifier and is trained on bootstrap samples of the training data.

Pseudocode of bagging
Random Forest
Random Forest6 is a versatile machine learning strategy equipped for performing both regression and Classification. Random Forest is appropriate when target protein is not decided or unknown, because random forest can find good combinations of features from many available features. This algorithm creates forest with number of trees as its name recommends. It is an ensemble method. Ensembles offer an effective technique for obtaining increased levels of predictions of many different learning algorithm instances. Ensemble methods provide better performance. RF is a form of ensemble machine learning algorithm called Bootstrap Aggregation or bagging. Bagging is known to lessen the variance of the algorithm. RF takes a subset of observations and subset of factors to build a decision tree. It fabricates different such decision tree and combine to get a more exact and stable prediction. The more trees in the forest, the more robust the forest resembles. Similarly in Random Forest classifier, the more number of trees in the forest gives the high precision. It handles missing values. Random Forest classifier won’t over fit the model, when we have more trees in the forest. Random forest can deal with huge dataset with higher dimensionality is its principle advantage. It has a powerful strategy for assessing missing information and keeps up precision when a huge extent of the information is missing. It has strategies for adjusting errors in data sets where classes are over-burden.
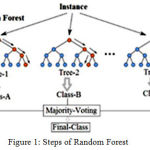 |
Figure 1: Steps of Random Forest.
|
Create random subset of features of training class with random values. We make decision trees. Again values will be changed and another decision tree will be created. This is why it is called forest. Next class prediction is to be done, vote for each observation and decide about class of observation based on the result.

Pseudo code of Random Forest
Decision Tree
Decision tree is an algorithm which is used for Classification. A decision tree is a series of decisions. Each of those results prompts to additional nodes, which expand into different conceivable outcomes. This gives it a treelike shape. It is generally illustrated as a tree, with the root at the top and leaves at the bottom. DT’s are mostly represented in top-down approaches. Decision tree is utilized in various fields such as in machine learning, data mining, and statistics. Decision trees are also known as classification trees in which nodes represent data rather than decisions. In DT each branch represents set of attributes or rules which is related to a specific class label, which is at the end of the branch.
Rotation Forest
A comparatively new ensemble learning method is Rotation Forest, which uses individually trained decision tree. To generate the base classifier’s training data the list of capabilities is haphazardly part into N subsets where N is the parameter of the calculation and Principle Component Analysis (PCA) is enforced to every subset. In order to identify patterns in data, PCA is used in paper.3 It’s a dynamic tool for analyzing data. Principle components are maintained in order to perpetuate the inconstancy data in the information. For a base classifier to form new features, K axis rotation takes place. PCA is a dimensionality reduction strategy in which a covariance analysis between factors happen. PCA is helpful when there is information on an extensive number of factors, and there is some redundancy in those factors.3 For this situation repetition implies that a portion of the factors are associated with each other. Linear Discriminant Analysis, Quadratic Discriminant Analysis transformations are also applied.

Pseudo code of Rotation Forest
Support Vector Machines
Most commonly used Machine learning algorithm is SVM. For handling high dimensional data SVM is used. In n-dimensional space every data point is sketched in the case of SVM.SVM depends on the idea of decision planes that characterize the decision limits. Classification is performed by finding the hyper plane and then the equation will be obtained. Then the data points will be separated into two parts points with same class label lie on one side and rest on the other side. Support vectors are points which are closer to hyper plane. Margin is referred as the distance between hyper plane and the nearby data point.
K-Nearest Neighbour
Classification and regression divining problems can be done using KNN. But it is more extensively used in classification problems in the industry. In KNN nearest neighbors are identified and using majority voting prediction is done. For identifying the neighbors a distance measure is used. The most suitable distance measure used is Euclidean distance. Euclidean distance is determined as the square root of the sum of the squared differences between a new point(x) and an existing point (x2) across all inputs attribute j. With the size of training dataset the computation complexity of KNN increases. KNN makes forecast by computing the likeness between an input data and each training occurrence.
Naive Bayes
Naïve Bayes algorithm7 is one of the probabilistic classifier. It relies upon probability models that coordinate solid independence assumptions. The independence assumptions routinely don’t influence reality. In this manner they are considered as Naïve. The Naive Bayes Classifier system depends on the so-called Bayesian theorem and is especially suited when the dimensionality of the data sources is high. Naive Bayes classifiers can deal with a subjective number of independent variables whether continuous or categorical. Depending on the possibility of the probability model, you can set up the Naive Bayes calculation in a managed learning setting. The benefit of utilizing the algorithm is that it is straightforward, and assemble brief than logistic regression. Another favorable position of the algorithm is that it functions admirably limited measures of training to calculate the analysis parameters.
Result and Analysis
Dataset
Data8,9 are collected from various sources which include PDB Drug Data Bank, PubChem. The compounds used for this analysis are mainly from medicinal plants. We selected important molecular descriptors and considered the molecular descriptor values and eight rules which are used in drug likeness prediction. Eight rules are Lipinski’s rule, Ghose filter rule, Verber’s rule, CMC-50 rule, MDDR rule, BBB Likeness, Weighted QED, and UnWeightedQED. 680 compounds were analysed and considered for finding out the properties and rules. A web based application is developed in which compound name can be selected and all the properties of the compound will be retrieved. Our main aim in this paper is to predict Drug likeness using Tree based Ensemble Classifier. With the invention of Virtual Screening Drug discovery process became easy. The training data set consists of three classes low, medium and high.
The implementation of Random Forest, KNN, SVM,10 Decision Tree, Naïve Bayes, Rotation Forest12 and a comparison of classification accuracy of all these algorithms are done. The best accuracy is given by Rotation Forest algorithm. A web based tool for the prediction of drug likeness of chemical compounds is done. The application is based on the Rotation Forest Classification algorithm. It uses the training data and test data from the data set already collected.
 |
Figure 2: Interface.
|
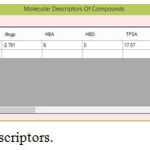 |
Figure 3: Molecular Descriptors.
|
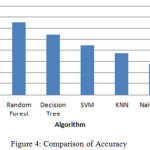 |
Figure 4: Comparison of Accuracy.
|
The above bar chart shows the variation in the accuracy of algorithms. According to this Fig3 Rotation Forest shows best accuracy. Table 1 shows the performance comparison between methods based on accuracy rate, precision, recall and F-measure.
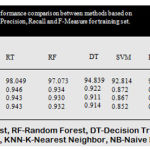 |
Table 1: Performance comparison between methods based on accuracy rate, precision, recall and F-measure for training set.
|
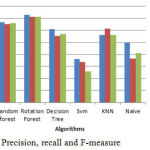 |
Figure 5: Precision, recall and F-measure.
|
Conclusion
Our paper focuses on prediction of Drug likeness based on machine learning methods. Finally comparisons between various algorithms are done in our work. We have done data collection based on ADMET properties11 and eight rules like Lipinski’s rule, Ghose filter rule, Verber’s rule, CMC-50 rule, MDDR rule, BBB Likeness, Weighted QED, and UnWeighted QED. The compounds identified for this work are mainly from medicinal plants. Based on the eight rules we have classified the compounds into two class labels which is categorised as Drug and non-Drug. We have implemented Random Forest, Decision Tree, SVM, Naïve Byes, Rotation Forest and KNN to have a comparison between its accuracy. Rotation Forest and Random Forest outperformed KNN, Decision Tree, SVM and Naïve Bayes. Rotation Forest showed better accuracy for prediction. Bagging in SVM and KNN are considered for the future work. Our future work also includes finding a rare disease and identifies the protein structure of the disease from PDB bind Database. Docking study with the screened set of compounds is to be done. Docking performs an essential role in Drug discovery. The transformation in the Rotation forest may be changed using other discriminant analysis methods like LDA, RLDA, ICA, CCA and QDA and a comparative study of these can be done in the future study.
References
- Selcuk K., Zararsiz G and Goksuluk D . Drug/nondrug classification using support vector machines with various feature selection strategies. computer methods and programs in biomedicine. 2014;2:51-60.
- Gaspar C., et al. Automatic selection of molecular descriptors using random forest: Application to Drug discovery. Expert Systems with Applications. 2017;72:151-159.
CrossRef - Juan R. J., Ludmila I. K and Carlos J. A. Rotation forest: A new classifier ensemble method. IEEE transactions on pattern analysis and machine intelligence. 2006;28:101619-1630.
- Ani R., Deepa O. S. Rotation Forest Ensemble Algorithm for the Classification of Phytochemicals from the Medicinal plants. Journal of Chemical and Pharmaceutical Science.
- Yongjun., et al. A new ensemble method with feature space partitioning for high-dimensional data classification. Mathematical Problems in Engineering. 2015;2015.
- Ani R., et al. Modified Rotation Forest Ensemble Classifier for Medical Diagnosis in Decision Support Systems. Progress in Advanced Computing and Intelligent Engineering. 2018. 137-146. Springer, Singapore.
- Antonio L. Machine-learning approaches in Drug discovery: methods and applications. Drug discovery today. 2015;20(3):318-331.
CrossRef - http://bioinfapplied.charite.de/supernatural_new/index.php?site= compound_input.
- http://www.niper.gov.in/pi_dev_tools/DruLiToWeb/DruLiTo_index.html.
- Kavitha K. R., et al. A correlation based SVM-recursive multiple feature elimination classifier for breast cancer disease using microarray. 2016 International Conference on Advances in Computing, Communications and Informatics (ICACCI). 2016:2677-2683.
- García-Sosa., Alfonso T., et al. Drug Logit logistic discrimination between Drugs and nondrug including disease-specificity by assigning probabilities based on molecular properties. Journal of chemical information and modeling. 2012;52(8 ):2165-2180.
- Thomas E. M., David J. B and Peter J. H. Virtual screening of Chinese herbs with random forest. Journal of chemical information and modeling. 2007;47(2):264-278.
CrossRef

