
Manuscript accepted on :31-July-2018
Published online on: 16-08-2018
Plagiarism Check: Yes
Reviewed by: Kulvinder Kaur
Second Review by: Abhinav Kanwal
I Made Pande Dwipayana1,2 , Karismayusa Sudjana3, Siswadi Semadi1, Ketut Suastika1, Made Ratna Saraswati1 and Wira Gotera1
, Karismayusa Sudjana3, Siswadi Semadi1, Ketut Suastika1, Made Ratna Saraswati1 and Wira Gotera1
1Division of Endocrinology and Metabolism, DepartmentofInternal Medicine, Facultyof Medicine, Udayana University-Sanglah General Hospital Denpasar Bali.
2Doctoral Programme in Medical Science, Facultyof Medicine, Udayana University Denpasar Bali.
3Internal Medicine Programme, Facultyof Medicine, Udayana University-Sanglah General Hospital Denpasar Bali.
Corresponding Author E-mail: pande_dwipayana@unud.ac.id
DOI : https://dx.doi.org/10.13005/bpj/1497
Abstract
We have reported a case of 21 year old patient with congenital adrenal hyperplasia that manifestated with ambiguous genitalia and other signs of androgen excess. Chromosome analysis revealed 46 XX. Laboratory examination and imaging showed high level of 17-hydroxyprogesterone, undeveloped uterus, two ovaries with follicles, no testicles, no prostate, and mass at upper side of both kidney with irregular border confirmed the diagnosis. It was planned to give glucocorticoid therapy to the patient to suppress androgen level, genital reconstruction surgery and psychosexual therapy to reared as a woman, but she refused all suggestions because she wanted to be considered a man.
Keywords
Androgen excess, Congenital Adrenal Hyperplasia, Genital reconstruction, Glucocorticoid therapy
Download this article as:| Copy the following to cite this article: Dwipayana I. M. P, Sudjana K, Semadi S, Suastika K, Saraswati M. R, Gotera W. Simple Virilization Type of Classic Congenital Adrenalhyperplasia: Case Report. Biomed Pharmacol J 2018;11(3). |
| Copy the following to cite this URL: Dwipayana I. M. P, Sudjana K, Semadi S, Suastika K, Saraswati M. R, Gotera W. Simple Virilization Type of Classic Congenital Adrenalhyperplasia: Case Report. Biomed Pharmacol J 2018;11(3). Available from: http://biomedpharmajournal.org/?p=21889 |
Introduction
Congenital adrenal hyperplasia (CAH) is a group of autosomal recessive genetic disorders. CAH is caused by deficiency of enzymes needed in adrenal steroid biosynthesis, this will result in increased production of adrenocorticotropic hormone (ACTH) and will further enlarge the adrenal gland. According to Office of Rare Diseases (ORD) from National Institutes of Health (NIH), CAH is a rare disorder, in United States of America the incidence is less than 200.000 of total population. Indonesia recorded 292 patients with CAH from 2009-2014, this data was gained by pediatric endocrinologists because no case has been detected in adult patients.1
Case Report
Patient wasa 21 year old, a Balinese, unmarried, consulted from Obstetric and Gynecology Department with ambiguous genitalia. Her urine flows from her women’s part of her genital, and she wanted to be able to urinate in standing position like a man. She has always felt that she is a man, but her family treated her as a girl since she was born. During childhood she had taller figure compared to her friends, but her growth has been slowed down since adolesence. She also easily got pimpleson her face. She had never ejaculated and also has never gotten menarche. Agreement to do and take picture of physical examination especially genital examination were given by herself. Physical examination revealed short stature 133 cm, normal vital signs, tanner I breasts, penile gland-like enlarged clitoris with 5×2 cmin size. There was no hymen found from rectal toucher examination. Urethra was under the clitoris, the labia majoralooked like scrotum, testicles were unpalpable, and there was vagina, 4 cm depth from vaginal sondage examination. Speculum examination cannot be performed because of the small size of vagina.
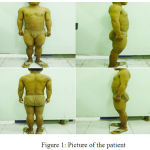 |
Figure 1: Picture of the patient
|
Laboratory examination revealed increase in 17-Hydroxyprogesterone (743. 53ng/dL), increase in dehydroepiandrosterone sulfate 667mcg/dL (80-350mcg/dL), normal range for testosterone and estradiol, decrease in folicle stimulating hormone 4.85mIU/mL (5-20mIU/mL), decrease in luteinizing hormone 1.45mIU/mL (5-20mIU/mL), and patient’s chromosome analysis result was 46 XX.
Trans abdominal ultrasonography and computed tomography showed undeveloped uterus, two ovaries with follicles, notesticles, noprostate, and massatupperside of both kidney with irregular border.
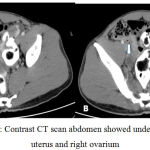 |
Figure 2: Contrast CT scan abdomen showed undeveloped uterus and right ovarium
|
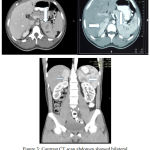 |
Figure 3: Contrast CT scan abdomen showed bilateral upper kidney mass.
|
We assessed this patient with congenital adrenal hyperplasia simple virilization type, primary amenorrhea, bilateral adrenal tumour probably myelolipoma, and transexualism. Our multi dicipline team decided to keep the female identity of the patient with glucorticoid, hormonal therapy, genital reconstruction, and supportive psychotherapy. How ever the patient refused our recommendation because she still wanted to be identified as male.
Discussion
CAHis a group of autosomal recessive genetic disorders. CAH caused by deficiency of enzymes needed in adrenal steroid biosynthesis;21-hydroxylase, 11β-hydroxylase, 17α-hydroxylase, 3β-hydroxysteroid dehydrogenase or P450 oxidoreductase.2,3
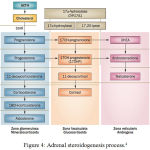 |
Figure 4: Adrenal steroidogenesis process
|
21-hydroxylase enzyme deficiency is the most common cause of CAH (90-95%).2,3,5 21-hydroxylase turns 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisol and progesterone to deoxycorticosterone, a precursor of cortisol and aldosterone.2,3 Steroidogenesis process in CAH can be viewed in picture 6. Failure of cortisol synthesis will increase ACTH from anterior pituitari and causes adrenal hyperplasia.6,7
There are two types of CAH, classic and non classic. The classic type consist of salt wasting type (75%) and simple virilization type (25%). In salt wasting type, there is no 21-hydroxylase, so both aldosterone and cortisol synthesis will be disrupted . In simple virilization type, a small part of the enzyme still can function normally so disruption only happens in cortisol synthesis.8,9
Clinical manifestations in CAH are caused by cortisol and aldosterone deficiency and excess of androgen production. Hormonal examination can help us to differentiate type of CAH, but the gold standard is molecular genetic analysis2-5,10
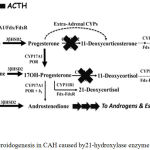 |
Figure 5: Steroidogenesis in CAH caused by21-hydroxylase enzyme deficiency.3
|
Table 1: Clinical and hormonal manifestation in CAH 11
| Manifes-tation | CAH type | |||||
| Salt Wasting | Simple Virilization | Non classic | ||||
| Male | Female | Male | Female | Male | Female | |
| Age whendiagnosed | Neonatus-6 month | Neonatus-1 month | 2 – 4 year | Neonatus-2 year | Children-adults | |
| External genital | Normal | Ambi-guous | Normal | Ambi-guous | Nor-mal | Normal/clitoro-megaly |
| Aldosterone | Low | Normal | Normal | |||
| Renin | High | Normal/increased | Normal | |||
| cortisol | Low | Low | Normal | |||
| 17-OHP | > 20.000 ng/dL | 10.000 – 20.000 ng/dL | ACTHStimulation: 1500 -10.000 ng/dL | |||
| 21-hydroxy-laseenzymeactivity | 0 | 1 – 10% | 10-75% | |||
In our case clinical manifestations are caused by androgen excess, such as short stature, ambiguous genital, acne, and amenorrhea. Our patient didn’t have hirsutism though, this can be caused by insensitivity of hair follicle to androgen, so the pilosebaceous unit differentiate to sebaceous gland.11,12,13
Treatment goals for CAH in adults are to prevent side effect from long term adrenal replacement therapy and to keep patient’s fertility.5,10,14,17 Modalities used in treatment can be seen at table 3. The corner stone of CAH treatment is glucocorticoid; hydrocortisone (15-45 mg/day) , prednisone (5-7.5 mg/ day), and dexamethasone (0.25-0.5 mg/day).9,10,12-15
Table 2: Medication used in CAH12
| Physiologic effects | Clinical effects | Side effects | |
| Glucocorticoid | Cortisol replacement therapy; suppressing hypothalamus-pituitari-adrenal axis to decrease adrenal androgen secretion. | Adrenal insuffiency therapy, prevention of virilization in women, treatment and prevention of infertility.
|
Iatrogenic cushing syndrome. |
| Fludrocortisone | Mineralocorticoid replacement therapy; suppresing renin-angiotensin-aldosterone axis, decreasing vasopressin, ACTH, and adrenal androgen secretion. | Keeping sodium and potassium balance, prevention of intravascular fluid depletion, decrease glucocorticoid dose.
|
Hypertension. |
| Contraception pill | Decrease serum androgen, suppressing hypothalamus-pituitari-adrenal axis to decrease ovarium androgen.
|
Regulate menstruation cycle, decrease hirsutism. | – |
| Spironolactone | Mineralocorticoid antagonis; competitive inhibitor of androgen receptor; inhibit 5a-reductase therefore decreasing androgen production. | Decrease hirsutism. | Diuresis. |
Fertility impairment in CAH cannot be treated only by steroid but also surgery for genital reconstruction and psychosexual therapy. Our patient refused the treatment recommendation to suppress androgen because she wanted to be identified as a man and wanted to do genital reconstruction surgery to be a man. Our team refused her request because currently there is no legal law about gender changing in Indonesia. Abundant androgen exposure since intrauterine period was very likely that contributed to her desire to be a man.
One of the complication from CAH is adrenal gland tumor, usually detected accidentally during imaging examination. This tumor is caused by long term high ACTH exposure. Our patient had non contrast enhancement tumor from her imaging, so we assumed that her tumor is myelolipoma.
Conclusion
We have reported case of patient with congenital adrenal hyperplasia that manifestated in ambiguous genitalia and other signs of androgen excess. Altough chromosome analysis revealed 46 XX, patient has always identified herself as a man because of her high level of androgen. Laboratory examination and imaging which resulted in high level of17-Hydroxyprogesterone, undeveloped uterus, two ovaries with follicles, no testicles, no prostate, and mass at upper side of both kidney with irregular border confirmed the diagnosis. Patient was planned to be given glucocorticoid therapy to suppress androgen level, genital reconstruction surgery and psychosexual therapy to be a woman, but she refused all suggestions because she wanted to be a man.
Conflict of Interest
There is no conflict of interest.
Funding Source
Author didn’t get any grant or funding for this article
References
- Rahmartani L.D. Mutasi gen CYP21 danprofilklinisanakndenganhiperplasia adrenal kongenital (tesis). Jakarta: Universitasindonesia. 2015.
- Speiser P.W, White P.C. Congenital Adrenal Hyperplasia. N Engl J Med 2003;349:776-788.
CrossRef - Auchus R.J. Management considerations for the adult with congenital adrenal hyperplasia. Molecular and Cellular Endocrinology. 2015;201:1-8.
CrossRef - Han T.S, Walker B.R, Arlt W, Ross R.J. Treatment and health outcomes in adults with congenital adrenal hyperplasia. Nat Rev Endocrinol 2014;10:115-124.
CrossRef - Speiser P.W, Azziz R, Baskin L.S, Ghizzoni L, Hensle T.W, Merke D.P, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J. Clin Endocrinol Metab. 2010;95:4133-4160.
CrossRef - Arlt W. Disorders of the adrenal cortex. In: Kasper D.L, Hauser S.L, Jameson J.L, Fauci A.S, Longo D.L, Loscalzo J, eds. Harrison’s principles of internal medicine. New York: Mc Graw Hill. 2015;2309-2329.
- Kurtoglu S, Hatipoglu N. Non-classical congenital adrenal hyperplasia in childhood. J.Clin Res Pediatr Endocrinol. 2017;9:1-7.
CrossRef - Dessinioti C, Katsambas A.D. Congenital adrenal hyperplasia. Dermato-Endocrinology. 2009;1(2):87-91.
CrossRef - Han T.S, Walker B.R, Arlt W, Ross R.J. Treatment and health outcomes in adults with congenital adrenal hyperplasia. Nat Rev Endocrinol. 2014;10:115-124.
- Ambroziak U, Bednarczuk T, Malinowska M.G, Malunowicz E.M, Gzechocinska B, Kaminski P, et al. Congenital adrenal hyperplasia due to 21-hydoxylase deficiency-management in adults. EndokrynologiaPolska/Polish. Journal of Endocrinology. 2010;61:142-155.
- White P.C, Speiser P.W. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocrine Reviews. 2000;21(3):245-291.
CrossRef - Merke D.P, Poppas D.P. Management of adolescents with congenital adrenal hyperplasia. Lancet Diabetes Endocrinol. 2013;1:341-352.
CrossRef - Hohl A, Ronsoni M.F, Oliveira M.D. Hirsutism: diagnosis and treatment. Arq Bras Endocrinol Metab. 2014;58(2):97-107.
CrossRef - Merke D.P. Approach to the adult with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J. Clin Endocrinol Metab. 2008;93(3):653-660.
CrossRef - Ogilvie C.M, Crouch N.S, Rumsby G, Creightont S.M, Liao L.M. Conway GS. Congenital adrenal hyperplasia in adults: A review of medical, surgical and psychological issues. Clinical endocrinology. 2006;64:2-11.
CrossRef

