
Manuscript accepted on :October 26, 2017
Published online on: --
Arunkumar T , Archana Vasuki K and Narendrakumar G
, Archana Vasuki K and Narendrakumar G
Department of Bioinformatics, School of Bio and Chemical Engineering, Sathyabama University, Chennai – 600045.
Corresponding Author E-mail: tarunkumart@gmail.com
DOI : https://dx.doi.org/10.13005/bpj/1307
Abstract
From the above results the natural plants and fruit extract containanti- bacterial properties to inhibit the action of disease caused by protein(Haemolysin) present in Vibrio species. To find the drug effectiveness and inhibit the action of disease caused by protein by natural compounds is done by ligand-receptor interactions,pharmacophore and ADMET studies. The ADMET studies were done for 10 natural compounds and 6 were satisfied. The common feature pharmacophore studies were done for 10 compounds (Poses A, B,C,D). The ligand -receptor interactionswere analysed for 60 ligands with common receptor (E,F,G,H,I,J,K,L,M,N). Molecular dynamics and simulation analysed for two natural compounds(Cyanidin) and (Bergapten).
Keywords
Haemolysin Molecular dynamics;pharmacophore; simulation; Vibrio paraheamolyticus;
Download this article as:| Copy the following to cite this article: Arunkumar T, Vasuki K. A, Narendrakumar G. In Silico Analysis on Docking Studies of Haemolysin Protein in Vibrio Paraheamolyticus. Biomed Pharmacol J 2017;10(4) |
| Copy the following to cite this URL: Arunkumar T, Vasuki K. A, Narendrakumar G. In Silico Analysis on Docking Studies of Haemolysin Protein in Vibrio Paraheamolyticus. Biomed Pharmacol J 2017;10(4). Available from: http://biomedpharmajournal.org/?p=17874 |
Introduction
Vibriosis is a disease caused by vibrio belongs to genus Vibriobacteria. It is a Gram-negative bacterium, possessing a curved-rod shape, Vibriosis is mainly causative disease for marine organism like shrimps, fishes etc.other than marine organism it will cause disease to humans by foodborne infection and is associated with eating undercooked seafood.1,2,3 Vibrio Bacterial diseases, have been reported in costal area and marine organism by 14 species in that strains Vibrio parahaemolyticusis an Asian strain found in sea water and coastal environments which is a more virulent and causes wound in humans, shrimps and fishes.Vibrio parahaemolyticus causing disease in fishes like tuna, marakkal, sardines It causes the wound, black lesion and even sudden death in fishes. it causes loss to many seafood companies and hatcheries.4,5,6,7
A protein secreted by Vibrio parahaemolyticus, is a type of hemolysin that is most virulent and acknowledged in past decades as the important pathogenic factor. It is differentiated into (trh) thermostable related haemolysin and (tdh) the thermoreliable direct haemolysin. Although originally studied for its hemolytic property, TDH has been long suspected to be an enterotoxin involved in most cases of Vibrio parahaemolyticus.8,9,10
Haemolysin is responsible of lysis of red blood cells in bacteria it also causes Hemolytic cancer in the human. The three types of heamolysin are α,β and γ.α–Haemolysin is mainly responsible for oligomerization of water-filled channels and toxin monomer in cell depolarization, osmotic phenomena and loss of vital molecules leading to its demise.β– Heamolysin is a toxic mechanism to be the hydrolysis of a specific membrane lipid causes cell lysis.γ-Haemolysin, it has a higher affinity for phosphocholines.11,12
The lytic process, most commonly seen in leucocytes, is caused by pore formation induced by an oligomerized octamer that organizes in a ring structure. Many studies show that naturally available compounds suppressor or inhibits the bacterial diseases (Vibriosis) caused by hemolysin.13,14,15,16 The main objective is to test the drug effectiveness to inhibit the action of the haemolysin (which is responsible for causing the disease) obtained from plant extracts, caffeine, curry leaves, flavonoids, grape seed, sweet basil, phenolic, papaya leaf extract compounds.
Materials and Methods
Molecular modeling of Hemolysin Protein.
To identify the function analysis of the protein hemolysine the sequence of haemolysin protein of α, β and γ sequences for Vibrio parahaemolyticuswas retrieved form the Uniprot database with uniprot ID P19250 for modelling because of unavailability of protein structure in the structural database. The homology structure was retrieved from structural database for the protein sequence the structure was predicted using a modeller 9.17.The modelled structure was analyzed using SAVS (Structure analysis and verification server) and MolProbity, is a structure-validation server that provides evaluation of model quality in global and local in basis of the arrangements of amino acids in Ramachandran plot for the modelled protein. For the predicted structure Energy minimization, Molecular dynamics simulation was performedusing Accelrys Discovery Studio (ADS) 2.0 for 1 nanosecond and stabilized structure model was used.
Identification of drug for Hemolysin Protein from natural plant compounds.
The natural drug compounds for hemolysin was identified in various research articles. The identified compound was collected from natural compounds like plant extracts, caffeine, curry leaves, flavonoids, grape seed, sweet basil, phenolic, papaya leaf extract compounds.10 naturally bioactive compounds were identified for hemolysin. Structural analogs for active pharmacologically compounds were retrieved from Pubchem database. Absorption, distribution, metabolism, elimination and toxicity (ADMET) were analyzed by Pharmacokinetic properties. Using Discovery Studio Receptor-ligand docking was done and the docked molecules are viewed for hydrogen bond interactions between the ligand atoms and the amino acid residues of the receptor molecule. The distance between the bonds is calculated and estimated for studying the favourable interactions between the ligand and the receptor molecule. Various interactions such as the alkyl bonds, pi bond, and the respective distance are calculated for the receptor distances are calculated the receptor-ligand complex. The ten compounds are
Molecular docking and Molecular dynamics studies on simulation on natural plant compounds
From the modeled hemolysine protein the selected drug compounds was docked by autodock by analyze using drug-likliness and ADMET properties. In ADMET the scores was measured through a multiple scoring functions like piecewise linear potential (‑PLP1), Ligand Score 1 and 2, and ‑PLP2, Jain, Ludi, Dock score, potentials of mean force, which results 6 compounds satisfy the ADMET Properties based on binding energy the docked compound was selected for the molecular dynamics and simulation studies.
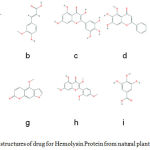 |
Figure 1-2D: structures of drug for Hemolysin Protein from natural plant compounds (a) Cyanidin, (b) Ferulic Acid, (c) Vanillic Acid, (d) Gallic Acid, (e) Myricetin, (f) Baicalein, (g) Pinocembrin, (h) Morin, (i)Ellagic acid, (j) Bergapten. |
Result and Discussion
Heamolysin
The non-structured target sequence were taken from uniprotKB .The similarity were checked in BLAST and the template structure 3A57A was retrieved from PDB. The sequences were modeled in MODELLER 9.17 server. In script file five DOPE score were generated among that lowest DOPE score was taken as protein structure and results will be executed in modeller window.
The target protein Haemolysin is modelled in modeller 9.17. Thecrystalline structure is viewed in Biovia 4.1.Modelled Protein Haemolysin (Fig 2).Molprobity result for target protein(haemolysin) (Fig 3)
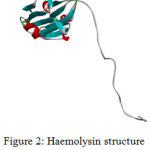 |
Figure 2: Haemolysin structure viewed in Biovia 4.1 |
 |
Figure 3: Ramachandran plot of haemolysin |
ADMET result analyses and interpretation
The seven natural compounds of Flavonoids, caffeine ,papaya leaf and grape seed are Subjected to ADMET (Adsorption Distubution, Metabolism, Toxicity studies) studies which gave 6 ligands satisfying the ADMET rules out of 10 compounds. The ADMET results for all the 6 compounds was shown in the given Table -1. (Fig.4).The plot highlights for BBB penetration levels Blue: Very high penetrant; High penetrant;cyan: Medium penetrant; Orange: Low penetrate.An optimal drug should not penetrate the BBB level as it can cause side effects in the CNS. Thus, drug compounds with BBB values 2 and 3 are considered optimal for a drug to be administered.
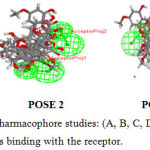 |
Figure 4: Common Feature Pharmacophore studies: (A, B, C, D) POSE 1,2,3,4 represents he different positions of ligands binding with the receptor. |
Table 1: ADMET Results Analyses
| S.NO | PubChem ID | BBB
Level |
Absorption
Level |
Solubility
Level |
Hepatoma
Toxicity |
CYP_2D | PPB
Level |
PSA_2D |
| 1 | 68247 | 4 | 0 | 3 | 1 | 1 | 1 | 116.631 |
| 2 | 445858 | 3 | 0 | 4 | 1 | 0 | 1 | 67.861 |
| 3 | 5281672 | 4 | 3 | 3 | 1 | 1 | 1 | 151.123 |
| 4 | 5281605 | 3 | 0 | 3 | 1 | 1 | 2 | 83.677 |
| 5 | 68071 | 2 | 0 | 4 | 1 | 0 | 2 | 67.861 |
| 6 | 8468 | 3 | 0 | 3 | 1 | 0 | 2 | 67.861 |
| 7 | 2355 | 2 | 0 | 3 | 1 | 0 | 1 | 47.715 |
| 8 | 70686876 | 3 | 0 | 4 | 1 | 1 | 2 | 82.766 |
| 9 | 370 | 3 | 0 | 3 | 1 | 0 | 1 | 100.562 |
| 10 | 5281855 | 4 | 1 | 3 | 1 | 0 | 1 | 135.723 |
Pharmacophore result interpretation
Common feature pharmacophore generation is studied for the ligand molecules obtained from headsetresult. It generates common feature pharmacopeia models from a set of ligands .it uses Hip-hop hypothesis to generate common feature pharmacophore among a set of active ligands. the algorithm can also optionally use information from inactive to place excluded volume features.The colours of the pharmacopeia are represented in given green, blue and magenta. The green denotes the hydrogen bond acceptors, blue represents the hydrophobic centres and Magenta represents hydrogen bond donors Figure -4.
Absolute energy
Absolute free energy calculations based on the thermodynamic cycle of a set of diverse inhibitors binding to a protein and demonstrating the mean absolute error of the protein or drug.
ConftNumber
ConftNunber or Converted Number is used to convert the structures into their corresponding 3D structure in Discovery Studio and convert interactions energy into pharmacophorepoints location and constraints.
Fit Value
Fit Value verses the negative logarithm of the activity while generating the 10 pharmacophoremodels were generated with corresponding statistical parameters such as cost values, RMSD and Fit Value.
Study of receptor ligand interactions
The receptor ligand interactions are studied for Ten natural compounds (Figure -5)
 |
Figure 5: Ligand-Receptor intreactions represents the receptor -ligands interactions of (a) Cyanidin ,(b) Ferulic acid, (c) Pinocembrin, (d) Myricetin, (e) Baicalein, (f) Vanilic Acid, (g) Galic Acid,(h) Bergapten,.(h) Morin, (j) Ellagic Acid. |
Above results for docking done by giving 10 and 4 poses for the ligand-receptor interactions as input. Out of the 61 compounds selected for docking 10 compounds generated successful results for receptor-ligand interactions with 84 poses. The Ligscore,PMF,PLP,Jain Score,Dock Score are the five values used to analyse the docking results. A higher score in all the above values shows a highly stable molecular interactions, Polar attractive interactions,Polar repulsive interactions, solvation of the protein and ligand and an entropy term for the ligand.
Solvation analysis of target protein
The variation of solvation entropy as a function of solute charge has been used to investigate hydrophobic and hydrophilic ordering and the structure-making and structure breaking effects of ion.The solvation energy, free energy, entropy, etc. are defined as the difference in these quantities for the box containing the water molecules and the solute molecule and two separate isolated boxes of volume V, one with the water molecules and one with the solute molecule.
Solvation result of target protein
Study of molecular dynamics and simulation:
The dynamics of the receptor are studied using standard cascade dynamics. Molecular dynamics and simulation analyses for cyanidin and bergapten Figure – 6.
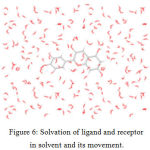 |
Figure 6: Solvation of ligand and receptor in solvent and its movement. |
Simulation result of kinetic energy graph for cyaniding
Molecular dynamics(MD) is a computer based method for the study of physical movements of atoms and molecules. The atoms and molecules can interact for a fixed period. The trajectories of atoms and molecules are determined by numerically solving Newton’s equation of motion. The PDB structure of Docked receptor-ligand is applied for Force field with CHARmm. Using Standard Dynamics method (SD) saves the current location of the coordinates from iteration to iteration. This method is useful for small changes, such as the removal of unfavourable steric contacts .Maximum steps: 2000,RMS:0.0001.Heating- A minimized structure represents the molecule at a temperature close to absolute zero. the equilibrium procedure is continued until various statistical properties of the system become independent of time. Steps:2000,time step:0.001.Save Frequent results-1000.
For the final molecular dynamics simulation, Charm takes the equilibrated structure as its starting point. In simulation, the trajectory traces the motions of the molecule through a period of at least several hundred picoseconds. Just as with energy minimization, provision is made to update the list of nonbonded interactions periodically. Additional options for handling nonbonded interactions and the environment are available, which makes the dynamics facility quite flexible.
Max step:1000000.Save frequent result -:true.
Simulation Graph shows (a)Kinetic energy vs time (b)Potential energy vs time (c)Total energy vs time mass required to do one work from rest to motion forbergapten (Figure -7)
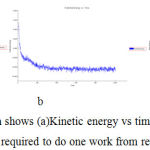 |
Figure 7: In simulation Graph shows (a)Kinetic energy vs time (b)Potential energy vs time (c)Total energy vs time mass required to do one work from rest to motion for cyanidin. |
Simulation result of kinetic energy graph for Bergapten
In simulation, the kinetic energy the mass required to do one work from rest to motion (Figure -8)
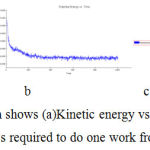 |
Figure 8: In simulation Graph shows (a)Kinetic energy vs time (b)Potential energy vs time (c)Total energy vs time mass required to do one work from rest to motion for bergapten. |
Conclusion
The natural plants and fruit extract containanti- bacterial properties to inhibit the action of disease caused by protein(Haemolysin) present in Vibrio species. The drug effectiveness and inhibit the action of disease caused by protein by natural compounds is done by ligand-receptor interactions,pharmacophore and ADMET studies.The ADMET studies were done for 10 natural compounds and 6 were satisfied. The common feature pharmacophore studies were done for 10 compounds receptor. Molecular dynamics and simulation is analysed for two natural compounds (Cyanidin) and (Bergapten) .During the simulation studies, these 2 compounds was shown better result.and found that there wasn’t much significant variation in the conformation. Hence, this identified compounds will be very much useful for to design novel and potential drug, which are
Considered to be an alternative compound for Hemolysin Protein.
References
- Inakollu S, Hung H.C and Shreve G.S. Biosurfactant enhancement of microbial degradation of various structural classes of hydrocarbon in mixed waste systems. Environ Eng Sci. 2004;21:463–469.
CrossRef - Nomura T. N. N and Matsumura M. The effect of copper concentration on the virulence of pathogenic Vibrio harveyi. Journal of Applied Microbiology. 2007;102(5):1300–1306.
CrossRef - Jayasree L, Janakiram P and Madhavi R. Characterization of Vibrio spp. Associated with Diseased Shrimp from Culture Ponds of Andhra Pradesh (India). Journal of the World Aquaculture Society. 2006;37:4 523.
CrossRef - Nakayama T, Nomura N and Matsumura M. Analysis of the relationship between luminescence and toxicity of Vibrio carchariae pathogenic to shrimp. Fisheries Science. 2005;71(6):1236.
CrossRef - Gopalakrishnan A and Parida A. Incidence of loose shell syndrome disease of the shrimp Penaeus monodon and its impact in the grow-out culture. Current Science. 2005;88:1148–1154.
- Lightner D.V. Diseases of cultured penaeid shrimp. In: J.P. McVey (ed.) CRC Handbook of Mariculture, Second edition,Crustacean Aquaculture. CRC Press Inc., Boca Raton, FL. 1993;1:393-486.
- Lavilla-Pitogo C.R, Leano E. M, Paner M.G. Mortalities of pond-cultured juvenile shrimp Penaeus monodon associated with dominance of luminescent vibrios in the rearing environment. Aquaculture. 1998; 164:337–349.
CrossRef - Adams A. Response of penaeid shrimp to exposure to Vibrio species. Fish Shellfish Immunol. 1991;1:59–70.
CrossRef - Phuoc L. H, Corteel M, Nauwynck H, Pensaert J, Alday-Sanz V, vander Broeck W, Sorgeloos P and Bossier P. Increased susceptibility of white spot syndrome virus infected Litopenaeus vannamei to Vibrio. Environ Microbiol. 2008;10:2718–2727.
- Gil A.I, Miranda H, Lanata C. F, Prada A, Hall E. R , Barreno C.M, et al. O3:K6 serotype of Vibrio parahaemolyticus identical to the global pandemic clone associated with diarrhea in Peru. Int J Infect Dis. 2007;11:324-8.
CrossRef - Zhou X, Konkel M.E and Call D. R. Type III secretion system 1 of Vibrio parahaemolyticus induces oncosis in both epithelial and monocytic cell lines. Microbiology. 2009;155:837-851.
CrossRef - Zhou X, Shah D.H, Konkel M.E and Call D.R. Type III secretion system 1 genes in Vibrio parahaemolyticus are positively regulated by ExsA and negatively regulated by Mol. Microbiol. 2008;69:747-764.
- Ralph A and Currie B.J. Vibrio vulnificus and V. parahaemolyticus necrotising fasciitis in fishermen visiting an estuarine tropical northern Australian location. J. Infect. 2007;54:e111-e114.
CrossRef - Abel B.R, Elwell C.S, van Ijzendoorn C.D and Engel J.N. 2004. Lipid raft-mediated entry is not required for Chlamydia trachomatis infection of cultured epithelial cells. Immun. 2004;72:7367-7373.
- Gurcel L, Abrami L, Girardin S, Tschopp J and van der Goot. F.G. 2006. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. 2006;126:1135-1145.
CrossRef - Hamada D.T, Higurashi K, Mayanagi T, Miyata T, Fukui T, Iida T.H and Yanagihara I. Tetrameric structure of thermostable direct hemolysin from Vibrio parahaemolyticus revealed by ultracentrifugation, small-angle X-ray scattering and electron microscopy. Mol. Biol. 2007;365:187-195.
CrossRef - Kumar R.B, Suresh M.X, Priya B.S. Pharmacophore modeling, in silico screening, molecular docking and molecular dynamics approaches for potential alpha-delta bungarotoxin-4 inhibitors discovery. Phcog Mag. 2015;11:19-28.
CrossRef - Lipinski C.A, Lombardo F, Dominy B.W, Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001; 46:3-26.22.
- Laurie A.T, Jackson R.M. Q-SiteFinder: An energy-based method for the prediction of protein-ligand binding sites. 2005;21:1908-16.23.
- Binkowski T.A, Naghibzadeh S,Liang J. CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 2003;31:3352-5.24.
- Peters K.P, Fauck J, Frömmel C. The automatic search for ligand binding sites in proteins of known three-dimensional structure using only geometric criteria. J Mol Biol. 1996;256(25):201-13.
- Morris G.M, Goodsell D.S, Halliday D.S, Huey R, Hart W.E, Belew R, et al., Automated docking using a Lamarckian genetic algorithm and empirical binding free energy. J. Comp Chem. 1998;19:1639-62.
- Thangapandian S, John S, Sakkiah S, Lee K.W. Molecular docking and pharmacophore filtering in the discovery of dual-inhibitors for human leukotriene A4 hydrolase and leukotriene C4 synthase. J. Chem Inf Model. 2011;51:33-44.35.
- Berk C, Sabbagh M. N. Successes andfailures for drugs in late-stage development for Alzheimer’s disease. Drugs Aging. 2013;30:783-92.
CrossRef - Arunkumar T, Feba A.E, Narendrakumar G. Docking studies of benzisoxazole analogues in white spot syndrome virus. Research Journal of Pharmacy and Technology. 2017;10(8):1-7.
- Pruthi V, Cameotra S.S. Rapid identification of biosurfactant producing strains using a cell surface hydrophobicity technique. Biotechnol Tech. 1997;11;671–674.
CrossRef

