
Manuscript accepted on :October 24, 2017
Published online on: --
Jun Yuan1, Wayne W Xu2, Jing Wang2, Zach Kubin3, Wei-Jie Fang4 and H. Fai Poon2
1Zhejiang Hisun Pharmaceutical Co., Ltd., Zhejiang, China.
2Innova Med Biotechnology Co., Ltd., Guangdong, China.
3Shanghai ChemPartner Co., Ltd., Shanghai, China.
4Institute of Drug and Pharmaceutical Analysis, College of Pharmaceutical Sciences, Zhejiang University, Zhejiang, China.
DOI : https://dx.doi.org/10.13005/bpj/1270
Abstract
Successful therapeutic commercialization requires the demonstration of efficacy and safety of a drug during clinical trials, as well as the commercial feasibility of drug production with consistent quality. Mitigating risk in these three areas is the key strategy for pharmaceutical development success. One of the most effective ways of risk mitigation during therapeutic development is to perform drugability assessments of the molecule. Drugability assessment studies facilitate our understanding of biotherapeutics, predict clinical outcomes, and provide rationales for molecular optimization. Better understanding of biotherapeutic drugability ensures the manufacturability, safety, and efficacy in clinical development. Therefore, drugability assessment is the key for successful biotherapeutic commercialization. Here, we reviewed current literature, and summarized the major durability studies of biotherapeutics.
Keywords
Drugability Commercialization Biotherapeutics;Manufacturability;
Download this article as:| Copy the following to cite this article: Yuan J, Xu W. W, Wang J, Kubin Z, Fang W, Poon H. F. Drugability Studies are Keys to the Successful Commercialization of Biotherapeutics. Biomed Pharmacol J 2017;10(4). |
| Copy the following to cite this URL: Yuan J, Xu W. W, Wang J, Kubin Z, Fang W, Poon H. F. Drugability Studies are Keys to the Successful Commercialization of Biotherapeutics. Biomed Pharmacol J 2017;10(4). Available from: http://biomedpharmajournal.org/?p=17529 |
Introduction
Successful therapeutic commercialization requires the demonstration of efficacy and safety of a drug in clinical trials, as well as the commercially feasibility for production of the drug with consistent quality. From novel drug discovery to blockbuster commercialization, risk mitigation through drugability assessment is the key strategy for pharmaceutical development success.1 The term drugability often refers to the accessibility, efficacy, and safety of a therapeutic molecule that meets clinical and commercial needs.2-5 Therefore, the drugability assessment of a molecule plays a significant role at an early stage of drug discovery. When it is performed properly, it can reduce the chance of expensive late-stage developmental failure.3,4,6,7 While many reviews have summarized the drugability of chemical therapeutics in literature,2,5,8-12 few summarize biotherapeutic drugability assessment from current literature. Therefore, we reviewed thecurrent literature, and summarized the durability studies of biotherapeutics, particularly those studies that involve monoclonal antibodies (Mab).
Drugability of Biotherapeutics
When compared to traditional chemical therapeutics, biological drugs show promise in better safety, efficacy, specificity, and extended half-life. However, the molecular structural complexity of biotherapeutics underlies a variety of challenges and limitations. Examples of challenges and limitations are: long development time, inefficient penetration of cell membranes, high cost of manufacturing, unwanted immune response, and poor stability.13,14 During the early stage of discovery, the drugability of a biotherapeutic molecule should be extensively evaluated to reduce the chance of late-stage developmental failure. Figure 1 shows the primary goals to be achieved and major analytical techniques that are performed to determine drugability during early stages of drug discovery. Manufacturability, safety, and efficacy are the three major aspects of drugability that need to be addressed in the early stages of drug discovery.
Manufacturability
Biotherapeutics undergo multiple chemical and physical stresses during the manufacturing process. These stresses potentially lead to modifications of the target protein drug affecting its safety and effectiveness.15 Manufacturing stress induced chemical modifications include oxidation, deamidation, peptides bond hydrolysis, etc.; and manufacturing stress induced physical modifications includes denaturation, precipitation, and aggregation, etc. Some of these induced modifications (e.g., aggregation, oxidation, glycosylation) can lead to heterogenicity and hence, significant adverse effects on the safety, efficacy, and pharmacokinetics/ pharmacodynamics (PK/PD) of the biotherapeutic drug15,16 Therefore, a quality assessment of stress induced modifications can provide necessary understanding of the physiochemical structures needed for lead candidates optimization.
Recent advances in proteomic techniques have enhanced understanding and prediction of protein modification resulting from bioprocesses.17,18,19,20 These technologies provide diverse databases and analysis software to provide valuable information for controlling manufacturing stress induced protein modification.17,18,21-23 Sequence- or structure-based bioinformatic prediction tools have been applied in recent studies.17,18 Table 1.1 presents the summary of available computational tools for structural predictions. Biopharmaceutical companies use the In Silico platform to assess stress-induced modification tendencies in the molecular structure of biotherapeutics. By selecting the optimal candidates via the In Silico platform, biotherapeutic molecules can be optimized to eliminate potential structural liabilities during manufacturing.17,24 Optimized candidates are then selected for in vitro analysis including safety, yield, preformulation, and biological activity.17,24
Safety
Immunogenicity is one of the most common side effects for biotherapeutics. While the principal cause of the immune response is divided, immunogenicity leads to reduced drug efficacy, altered drug clearance, and shortened plasma half-life of the administered drug.25-28 Therefore, immunogenicity assessment is one of the most important principal safety evaluations for biotherapeutics in development.26,28
Immunogenic response can be divided into T-cell dependent and T-cell independent responses. In T-cell independent immunogenic response, B cells bind to the therapeutic protein and respond by transiently producing IgM antibodies.29 In T-cell dependent immunogenic response, the complex interplay among antigen presenting cells, T cells, secreted cytokines, and B cells occur in respond to the administered proteins.30 Thus, anti-drug antibodies (ADA) are usually generated by T-cell dependent immunogenic responses.25,30 Immunogenicity is one of the key risk attributes for clinical safety, so it is desirable to assess the potential immunogenic issues during the early discovery stages. A drug molecule should be redesigned if it is found to be highly immunogenic. One way to reduce the immunogenicity is by removing the T-cell epitopes of the biotherapeutic drug.24 It is feasible to predict the immunogenicity of the T-cell epitopes based on the amino acid sequence from In Silico because their core residues are limited to 9–10 amino acids.30 There are a number of computational tools for T-cell epitope prediction and assessment for potential immunogenicity evaluations.30-32 These In Silico predictions are usually followed by in vitro and ex vivo cell-based assays validation. Major histocompatibility complex/human leukocyte antigen (HLA/MHC) binding assays are common cell-based assays used to validate the In Silico predictions by measuring the affinity of predicted epitope to HLA in vitro.30,33-35 A series of T cells will then be used to further assess the immunogenicity, including cytokine response assays, proliferation assays, naïve human PBMC response assays etc.30,33-35 These in vitro assays also enable bioprocess related changes to be evaluated, such as antibody expression, post-translational modifications, and formulations etc.30 In vivo experiments with “humanized” animal models are also performed for immunogenicity testing of therapeutic candidates to further validate the findings in cell-based assays.36 The data generated from the in vitro and in vivo validation assays provides an important assessment to the immunogenicity of the biotherapeutic protein to reduce clinical safety risks.30,32
Typical screening assay strategies for immunogenicity of biopharmaceuticals are summarized in Table 2. The data derived from these immunogenicity-based assessments can be used to prioritize candidates for development when multiple candidates are available. These assessments can also be used to optimize biopharmaceutical candidates for cost-effective drug development and safety improvement via molecular redesign. Humanization is commonly used to significantly reduce immunogenicity.37-39 Moreover, physicochemical modification and aggregation elimination of therapeutics can also be used to reduce immunogenicity.40 Other immunogenicity reduction strategies such as PEGylation have been explored.41 However, studies show that anti-PEG antibodies have been caused by PEGylated biotherapeutics.42-46 Nevertheless, immunogenicity reducing technologies are a key to mitigating the clinical safety risks of biotherapeutic developments.
Efficacy
Efficacy is frequently linked to the pharmacokinetics (PK) of a drug because of the direct relationship between efficacy and dosage. Special considerations are applied to biotherapeutics because of their complex structures.47-49 The primary PK determinant of a biotherapeutic is their FcRn-mediated recycling. However, other factors, such as glycosylation, and target mediated drug disposition (TMDD), and anti-drug antibody (ADA) response can also have tremendous influence on its PK.47 Figure 3 illustrates the general structure of lgG1 and the factors that influence PK properties. PK data is important to reference during biotherapeutic structural optimization to achieve desirable exposure, safety, and efficacy profiles.47,49-51 The bioanalytical methods used for protein quantification during PK studies are mainly based on immunoassay methodologies. However, mass spectrometer based quantification techniques, including the bottom-up and top-down approach, are gaining popularity recently due to their advantages in development time, specificity, and throughput.52,53 In addition, fluorescent or radioactive labeled molecules are also frequently used to test absorption and distribution of Mab related therapeutics.54,55 The PK characteristics of biotherapeutics show differences from that of chemical therapeutics (Table 3). PK data of biologics such as Cmax, Area Under Curve (AUC), half-life, volume of distribution, and clearance can help provide significant insight into their stability in blood, potential nonlinearity of PK, as well as target specific distribution assessment.47,48,53 PK data from animal studies provides the basis of extrapolation for first-in-human (FIH) dosage information in clinical studies. Linear PK, target-mediated drug disposition (TMDD), and physiology-based pharmacokinetic (PBPK) models are the most common FIH quantitative models used in order to show proper efficacy in clinical studies.56
Linear Pharmacokinetic (PK) Models – Protein biotherapeutics exhibit linear PK after saturation of target. PK data obtained in preclinical animal studies are typically extrapolated by simple mathematical models describing mono-exponential, bi-exponential, or multiexponential profiles observed in plasma or serum exposure. By using allomeric scaling techniques, intended dosage for human exposure predicted.57
Target-Mediated Drug Disposition (TMDD) Models – TMDD is applied when a significant fraction of a drug binds to its pharmacological target with high affinity such that this interaction influences the distribution and elimination of the drug.58 A well-developed TMDD model can help predict drug-related (e.g., drug elimination and distributional rate constants) and target-related (e.g., receptor expression) parameters, and estimate the in vivo receptor occupancy.56,58
Physiology-based pharmacokinetic (PBPK) models – PBPK models describe physiological characteristics such as lymph flow, organ distribution, FcRn binding, and relationships with serum and tissue. In PBPK models, physiological parameters and the ADME data are integrated to represent a quantitative framework for mechanistic translation across species. This approach provides a starting point to evaluate the impact of drug-dependent properties and system-dependent properties on human PK profiles of biotherapeutics.56,59-61 Although it was first developed for chemical therapeutics, PBPK models have been extensively applied to biotherapeutics.59-61
Summary
Drugability of biotherapeutics is an integral concept in drug discovery. It involves the analyzation of a biological drugs’ structural and physicochemical properties, biological activity, pharmokinetics and toxicity. Drugability assessment studies facilitate our understanding of biotherapeutics, predict clinical outcomes, and provide rationales for molecular optimization. Nevertheless, many studies, such as PK drivers of efficacy and toxicity, immunogenicity response, PK extrapolation to humans, and exposure–response relationships in patients etc, are currently in progress to gain better understanding of biotherapeutic drugability in order to ensure the manufacturability, safety, and efficacy in clinical development. With new advances in modern biotechnology, drugability assessments are the key for successful biotherapeutic commercialization.
Table 1: Prediction Database for PTMs
| Database | Source |
| Scansite | http://scansite.mit.edu/ |
| PREDIKIN | http://predikin.biosci.uq.edu.au/pkr/ |
| NetPhos | http://www.cbs.dtu.dk/services/NetPhos/ |
| NetPhos | http://www.cbs.dtu.dk/services/NetPhosK/ |
| Big-PI-prediction | http://mendel.imp.ac.at/sat/gpi/gpi_server.html |
| GlycoMod | http://expasy.ch/tools/glycomod/ |
| NetOGlyc | http://cbs.dtu.dk/services/NetOGlyc/ |
| NetNGlyc | http://cbs.dtu.dk/services/NetNGlyc/ |
| DictyOGlyc | http://cbs.dtu.dk/services/DictyOGlyc/ |
| YinOYang | http://cbs.dtu.dk/services/YinOYang/ |
| Sulfinator | http://www.expasy.org/tools/sulfinator |
| Oglyc | http://www.expasy.org/tools/sulfinator |
Table 2: Screening Assay Strategies for Immunogenicity
| Physiochemical characterization | |
| In silico | |
| – T cell epitope predictions | |
| – B cell epitope predictions | |
| HLA/MHC binding assays | |
| T cell responses | |
| In vivo models |
Table 3: PK Characteristics Comparison of Chemical Therapeutics and Biotherapeutics.
| Attributes | Chemical Therapeutics | Biotherapeutics |
| Binding | Nonspecific | Specific |
| PK/PD | PK usually independent of PD; short half‐life |
PK usually dependent on PD; long half‐life; |
| Dose Proportionality |
Linear PK at low doses (usually therapeutic doses); nonlinear PK at high doses (after saturation of metabolic enzymes) |
Nonlinear PK at low doses; linear PK at high doses after saturation of target |
| Distribution | High volume of distribution | Distribution usually limited to blood and interstitial spaces |
| Metabolism | Metabolism by cytochrome P450 or other phase I/ phase II enzymes |
Catabolism by proteolytic degradation |
| Excretion | Typically, biliary and renal excretion |
No renal CL of intact antibody. May be cleared by damaged kidneys. Uncommon if MW >20 kDa |
| Immunogenicity | Not seen | Maybe seen |
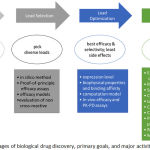 |
Figure 1: Stages of biological drug discovery, primary goals, and major activities
|
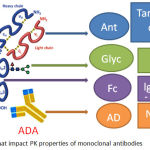 |
Figure 2: Factors that impact PK properties of monoclonal antibodies
|
Reference
- McCormick C.G, Henningfield J.E, Haddox J.D, Varughese S, Lindholm A, Rosen S, et al. Case histories in pharmaceutical risk management. Drug Alcohol Depend. 2009;105(Suppl1):42-55.
CrossRef - Di L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015;17(1):134-43.
CrossRef - Lugovskoy A.A, Reichert J.M, Beck A. 7th annual European Antibody Congress 2011: November 29-December 1, 2011, Geneva, Switzerland. MAbs. 2012;4(2):134-52.
CrossRef - Grigoriadis D.E, Hoare S.R, Lechner S.M, Slee D.H, Williams J.A. Drugability of extracellular targets: discovery of small molecule drugs targeting allosteric, functional, and subunit-selective sites on GPCRs and ion channels. Neuropsychopharmacology. 2009;34(1):106-25.
CrossRef - Egner U, Hillig R.C. A structural biology view of target drugability. Expert Opin Drug Discov. 2008;3(4):391-401.
CrossRef - Made V, Els-Heindl S, Beck-Sickinger A.G. Automated solid-phase peptide synthesis to obtain therapeutic peptides. Beilstein J Org Chem. 2014;10:1197-212.
CrossRef - Viswanadhan V.N, Balan C, Hulme C, Cheetham J.C, Sun Y. Knowledge-based approaches in the design and selection of compound libraries for drug discovery. Curr Opin Drug Discov Devel. 2002;5(3):400-6.
- Kerns E. D.L. Drug-like Properties: Concepts, structure design and methods from ADME to toxicity optimization: Elsevier. 2008.
- Basavaraj S, Betageri G.V. Can formulation and drug delivery reduce attrition during drug discovery and development-review of feasibility, benefits and challenges. Acta Pharm Sin B. 2014;4(1):3-17.
CrossRef - Lipinski C.A. Drug-like properties and the causes of poor solubility and poor permeability. J Pharmacol Toxicol Methods. 2000;44(1):235-49.
CrossRef - Gladki A, Kaczanowski S, Szczesny P, Zielenkiewicz P. The evolutionary rate of antibacterial drug targets. BMC Bioinformatics. 2013;14:36.
CrossRef - Jiang Z, Shao J, Yang T, Wang J, Jia L. Pharmaceutical development, composition and quantitative analysis of phthalocyanine as the photosensitizer for cancer photodynamic therapy. J Pharm Biomed Anal. 2014;87:98-104.
CrossRef - Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem. Inf. Model. 2012;52(2):360-72.
CrossRef - Sarkar A, Brenk R. To Hit or Not to Hit, That Is the Question – Genome-wide Structure-Based Druggability Predictions for Pseudomonas aeruginosa Proteins. PLoS One. 2015;10(9):e0137279.
CrossRef - Shire S.J. Formulation and manufacturability of biologics. Curr Opin Biotechnol. 2009;20(6):708-14.
CrossRef - Kamath K.S, Vasavada M.S, Srivastava S. Proteomic databases and tools to decipher post-translational modifications. J Proteomics. 2011;75(1):127-44.
CrossRef - Obrezanova O, Arnell A, dela C.R.G, Berthelot M.E, Gallagher T.R, Zurdo J, et al. Aggregation risk prediction for antibodies and its application to biotherapeutic development. MAbs. 2015;7(2):352-63.
CrossRef - Almagro J.C, Teplyakov A, Luo J, Sweet R.W, Kodangattil S, Hernandez-Guzman F, et al. Second antibody modeling assessment (AMA-II). Proteins. 2014;82(8):1553-62.
CrossRef - Poon H.F, Abdullah L, Reed J, Doore S.M, Laird C, Mathura V, et al. Improving image analysis in 2DGE-based redox proteomics by labeling protein carbonyl with fluorescent hydroxylamine. Biol Proced Online. 2007;9:65-72.
- Poon H.F, Calabrese V, Calvani M, Butterfield D.A. Proteomics analyses of specific protein oxidation and protein expression in aged rat brain and its modulation by L-acetylcarnitine: insights into the mechanisms of action of this proposed therapeutic agent for CNS disorders associated with oxidative stress. Antioxid Redox Signal. 2006;8(3-4):381-94.
CrossRef - Poon H.F, Castegna A, Farr S.A, Thongboonkerd V, Lynn B.C, Banks W.A, et al. Quantitative proteomics analysis of specific protein expression and oxidative modification in aged senescence-accelerated-prone 8 mice brain. Neuroscience. 2004;126(4):915-26.
CrossRef - Poon H.F, Hensley K, Thongboonkerd V, Merchant M.L, Lynn B.C, Pierce W.M, et al. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice–a model of familial amyotrophic lateral sclerosis. Free Radic Biol Med. 2005;39(4):453-62.
CrossRef - Poon H.F, Shepherd H.M, Reed T.T, Calabrese V, Stella A.M, Pennisi G, et al. Proteomics analysis provides insight into caloric restriction mediated oxidation and expression of brain proteins associated with age-related impaired cellular processes: Mitochondrial dysfunction, glutamate dysregulation and impaired protein synthesis. Neurobiol Aging. 2006;27(7):1020-34.
CrossRef - Zurdo J, Arnell A, Obrezanova O, Smith N, Gomez de la Cuesta R, Gallagher T.R, et al. Early implementation of QbD in biopharmaceutical development: a practical example. Biomed Res Int. 2015;2015:605427.
CrossRef - Baker M, Carr F. Pre-clinical considerations in the assessment of immunogenicity for protein therapeutics. Curr Drug Saf. 2010;5(4):308-13.
CrossRef - Wadhwa M, Thorpe R. The challenges of immunogenicity in developing biosimilar products. IDrugs. 2009;12(7):440-4.
- Kuriakose A, Chirmule N, Nair P. Immunogenicity of Biotherapeutics: Causes and Association with Posttranslational Modifications. J Immunol Res. 2016;2016:1298473.
CrossRef - Rup B, Pallardy M, Sikkema D, Albert T, Allez M, Broet P, et al. Standardizing terms, definitions and concepts for describing and interpreting unwanted immunogenicity of biopharmaceuticals: recommendations of the Innovative Medicines Initiative ABIRISK consortium. Clin Exp Immunol. 2015;181(3):385-400.
CrossRef - Sauerborn M, van de Vosse E, Delawi D, van Dissel J.T, Brinks V, Schellekens H. Natural antibodies against bone morphogenic proteins and interferons in healthy donors and in patients with infections linked to type-1 cytokine responses. J Interferon Cytokine Res. 2011;31(9):661-9.
CrossRef - Jawa V, Cousens L.P, Awwad M, Wakshull E, Kropshofer H, DeGroot A.S. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clin Immunol. 2013;149(3):534-55.
CrossRef - Walle V.I, Gansemans Y, Parren P.W, Stas P, Lasters I. Immunogenicity screening in protein drug development. Expert Opin Biol Ther. 2007;7(3):405-18.
CrossRef - Zurdo J. Developability assessment as an early de-risking tool for biopharmaceutical development. Pharmaceutical Bioprocessing. 2013;1(1):29-50.
CrossRef - McMurry J.A, Gregory S.H, Moise L, Rivera D, Buus S, Groot D.A.S. Diversity of Francisella tularensis Schu4 antigens recognized by T lymphocytes after natural infections in humans: identification of candidate epitopes for inclusion in a rationally designed tularemia vaccine. Vaccine. 2007;25(16):3179-91.
CrossRef - Kessler J.H, Mommaas B, Mutis T, Huijbers I, Vissers D, Benckhuijsen W.E, et al. Competition-based cellular peptide binding assays for 13 prevalent HLA class I alleles using fluorescein-labeled synthetic peptides. Hum Immunol. 2003;64(2):245-55.
CrossRef - Kessler J.H, Benckhuijsen W.E, Mutis T, Melief C.J, vander S.B.H, Drijfhout J.W. Competition-based cellular peptide binding assay for HLA class I. Curr Protoc Immunol. 2004;Chapter 18:Unit 18 2.
- Brehm M.A, Cuthbert A, Yang C, Miller D.M, DiIorio P, Laning J, et al. Parameters for establishing humanized mouse models to study human immunity: analysis of human hematopoietic stem cell engraftment in three immunodeficient strains of mice bearing the I L2rgamma(null) mutation. Clin Immunol. 2010;135(1):84-98.
CrossRef - Groot D.A.S, Terry F, Cousens L, Martin W. Beyond humanization and de-immunization: tolerization as a method for reducing the immunogenicity of biologics. Expert Rev Clin Pharmacol. 2013;6(6):651-62.
CrossRef - Roque-Navarro L, Mateo C, Lombardero J, Mustelier G, Fernandez A, Sosa K, et al. Humanization of predicted T-cell epitopes reduces the immunogenicity of chimeric antibodies: new evidence supporting a simple method. Hybrid Hybridomics. 2003;22(4):245-57.
CrossRef - Pavlinkova G, Colcher D, Booth B.J, Goel A, Wittel U.A, Batra S.K. Effects of humanization and gene shuffling on immunogenicity and antigen binding of anti-TAG-72 single-chain Fvs. Int J Cancer. 2001;94(5):717-26.
CrossRef - Kumar S, Singh S.K, Wang X, Rup B, Gill D. Coupling of aggregation and immunogenicity in biotherapeutics: T- and B-cell immune epitopes may contain aggregation-prone regions. Pharm Res. 2011;28(5):949-61.
CrossRef - Basu A, Yang K, Wang M, Liu S, Chintala R, Palm T, et al. Structure-function engineering of interferon-beta-1b for improving stability, solubility, potency, immunogenicity, and pharmacokinetic properties by site-selective mono-PEGylation. Bioconjug Chem. 2006;17(3):618-30.
CrossRef - Le Y, Toyofuku W.M, Scott M.D. Immunogenicity of murine mPEG-red blood cells and the risk of anti-PEG antibodies in human blood donors. Exp Hematol. 2017;47:36-47 e2.
- Povsic T.J, Lawrence M.G, Lincoff A.M, Mehran R, Rusconi C.P, Zelenkofske S.L, et al. Pre-existing anti-PEG antibodies are associated with severe immediate allergic reactions to pegnivacogin, a PEGylated aptamer. J Allergy Clin Immunol. 2016;138(6):1712-5.
CrossRef - Poppenborg S.M, Wittmann J, Walther W, Brandenburg G, Krahmer R, Baumgart J, et al. Impact of anti-PEG IgM antibodies on the pharmacokinetics of pegylated asparaginase preparations in mice. Eur J Pharm Sci. 2016;91:122-30.
CrossRef - Tung H.Y, Su Y.C, Chen B.M, Burnouf P.A, Huang W.C, Chuang K.H, et al. Selective Delivery of PEGylated Compounds to Tumor Cells by Anti-PEG Hybrid Antibodies. Mol Cancer Ther. 2015;14(6):1317-26.
CrossRef - Saifer M.G, Williams L.D, Sobczyk M.A, Michaels S.J, Sherman M.R. Selectivity of binding of PEGs and PEG-like oligomers to anti-PEG antibodies induced by methoxyPEG-proteins. Mol Immunol. 2014;57(2):236-46.
CrossRef - Liu L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell. 2017.
- Higel F, Seidl A, Sorgel F, Friess W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur J Pharm Biopharm. 2016;100:94-100.
CrossRef - Liu L. Antibody glycosylation and its impact on the pharmacokinetics and pharmacodynamics of monoclonal antibodies and Fc-fusion proteins. J. Pharm Sci. 2015;104(6):1866-84.
CrossRef - Feng Y, Zhao Q, Chen W, Wang Y, Crowder K, Dimitrov D.S. A new bispecific antibody targeting non-overlapping epitopes on IGF2: design, in vitro characterization and pharmacokinetics in macaques. Exp Mol Pathol. 2014;97(3):359-67.
CrossRef - Avery L.B, Wang M, Kavosi M.S, Joyce A, Kurz J.C, Fan Y.Y, et al. Utility of a human FcRn transgenic mouse model in drug discovery for early assessment and prediction of human pharmacokinetics of monoclonal antibodies. MAbs. 2016;8(6):1064-78.
CrossRef - Bronsema K.J, Bischoff R, vande N.M.C. Internal standards in the quantitative determination of protein biopharmaceuticals using liquid chromatography coupled to mass spectrometry. J. Chromatogr B Analyt Technol Biomed Life Sci. 2012;893-894:1-14.
CrossRef - Anderson L, Hunter C.L. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5(4):573-88.
CrossRef - Oeste C.L, Seco E, Patton W.F, Boya P, Perez-Sala D. Interactions between autophagic and endo-lysosomal markers in endothelial cells. Histochem Cell Biol. 2013;139(5):659-70.
CrossRef - Romer T, Leonhardt H, Rothbauer U. Engineering antibodies and proteins for molecular in vivo imaging. Curr Opin Biotechnol. 2011;22(6):882-7.
CrossRef - Wang J, Iyer S, Fielder P.J, Davis J.D, Deng R. Projecting human pharmacokinetics of monoclonal antibodies from nonclinical data: comparative evaluation of prediction approaches in early drug development. Biopharm Drug Dispos. 2016;37(2):51-65.
CrossRef - Lee J.W. ADME of monoclonal antibody biotherapeutics: knowledge gaps and emerging tools. Bioanalysis. 2013;5(16):2003-14.
CrossRef - Levy G. Pharmacologic target-mediated drug disposition. Clin Pharmacol Ther. 1994;56(3):248-52.
CrossRef - Davda J.P, Jain M, Batra S.K, Gwilt P.R, Robinson D.H. A physiologically based pharmacokinetic (PBPK) model to characterize and predict the disposition of monoclonal antibody CC49 and its single chain Fv constructs. Int Immunopharmacol. 2008;8(3):401-13.
CrossRef - Shah D.K, Betts A.M. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67-86.
CrossRef - Heiskanen T, Heiskanen T, Kairemo K. Development of a PBPK model for monoclonal antibodies and simulation of human and mice PBPK of a radiolabelled monoclonal antibody. Curr Pharm Des. 2009;15(9):988-1007.
CrossRef

